
ABSTRACT
Small non-coding RNAs play a pivotal role in gene regulation, repression of transposable element and viral activity in various organisms. Among the various categories of these small non-coding RNAs, microRNAs (miRNAs) guide post-translational gene regulation in cellular development, proliferation, apoptosis, oncogenesis, and differentiation. Here, we performed a genome-wide computational prediction of miRNAs to improve the understanding of miRNA observation and function in molluscs. As an initial step, hundreds of conserved miRNAs were predicted in 35 species of molluscs through genome scanning. Afterwards, the miRNAs’ population, isoforms, organization, and function were characterized in detail. Furthermore, the key miRNA biogenesis factors, including AGO2, DGCR8, DICER, DROSHA, TRABP2, RAN, and XPO5, were elucidated based on homologue sequence searching. We also summarized the miRNAs’ function in biomineralization, immune and stress response, as well as growth and development in molluscs. Because miRNAs play a vital role in various lifeforms, this study will provide insight into miRNA biogenesis and function in molluscs, as well as other invertebrates.
Introduction
Small RNAs, < 200 nucleotide (nt) in length, are usually non-coding RNA molecules that play a pivotal role in the regulation of gene expression at the post-transcriptional level [Citation1–3]. Small RNAs can be divided into classes based on their size and biogenesis partner proteins of the Argonaute family [Citation4,Citation5]. The main categories include microRNAs (miRNAs), endogenous small interfering RNAs (endo-siRNAs), and PIWI-interacting RNAs (piRNAs) [Citation6]. In fact, miRNAs and endo-siRNAs are generated from double-stranded precursors by DICER and are of 20–23 nt in length, while piRNAs are generated from single-stranded precursors and have a length of 26–31 nt, which occurs independently of RNase III enzymes. These enzymes are, in contrast, necessary for miRNA and endo-siRNA biogenesis. Among these three classes, miRNAs are generated from local hairpin structures by the action of two RNase III enzymes, DROSHA and DICER [Citation7].
Since the first discovery of the miRNA family, i.e., lin-4 and let-7, in Caenorhabditis elegans in 1993 [Citation8,Citation9], a large number of endogenous non-encoded miRNAs have been identified. Currently, 38,589 hairpin precursors and 48,860 mature miRNAs have been discovered in 271 organisms, from simple multicellular organisms to mammals, and these have been deposited in the public available miRNA database miRBase 22.0 [Citation10]. The majority of miRNAs are conserved in both sequence and function in Metazoa. Over half of C. elegans’s miRNAs share sequence homology with the miRNAs encoded in both fly and human genomes, which indicates that miRNAs are extensively conserved throughout animals’ evolution [Citation11]. These miRNAs can also be excellent phylogenetic markers for use in solving the problems of Metazoa macro-evolution [Citation12,Citation13].
The best-known function for miRNAs is the repression of gene expression, which is involved in cellular development, proliferation, apoptosis, oncogenesis, differentiation, and disease [Citation5,Citation14,Citation15]. A previous study reported that up to 30% of human protein-coding genes may be regulated by miRNAs [Citation16]. In most cases, the 3ʹ UTR of the target mRNAs is bound by the miRNA through imperfect complementary base pairing, which results in mRNA destabilization or translational repression [Citation17]. Tens of thousands of documented cases of targets down-regulated by miRNAs have bene found, while a handful of cases of miRNA-guided up-regulation associated with micro-ribonucleoproteins (microRNPs) have been observed in Xenopus laevis and humans [Citation18,Citation19]. Both miRNAs and the co-factors, such as AGO and DICER protein, have been detected in mammalian cell nuclei and are believed to directly regulate the promoter of the target gene via methylation [Citation20–22].
Molluscs are the second largest phylum in Metazoa and are widely distributed in the world. As successful colonizers of oceanic, limnetic, and intertidal zones, molluscs are remarkably resilient to harsh environmental conditions, including wide ranges of salinity, the hydraulic pressure of the deep sea, huge fluctuations in temperature, and prolonged air exposure [Citation23]. Thousands of genes are involved in the immune and stress responses, and these interact in a complex way, with many gene families having been expanded to reveal high sequence, structural, and functional diversity [Citation24]. Very few studies have addressed the diversity and function of miRNAs in molluscs. To improve the understanding of miRNA characteristics and function in molluscs, we performed a genome-wide scanning of miRNAs and functional analysis based on 35 molluscs species in the present study.
Results
Genome-wide scanning of miRNAs in molluscs
To explore the evolution of the miRNA repertoire in molluscs (), we devised an algorithm for finding homologs of conserved miRNAs based on whole-genome scanning and alignment. Here, hundreds of miRNAs were identified, following specific scanning criteria for the genome of each mollusc species (). The details of the scanned miRNA for each species were listed in the supplementary materials section under Table S1 and Fig. S1. The number of identified miRNAs increased proportionately with genome size, whereas cephalopods have a low density of predicted genomic miRNA (). The genomic miRNA density was defined as the ratio of the number of predicted miRNAs and the genome size (Mb). The predicted miRNAs were classified into miRNA families based on the miRBase 22 miRFam database. Among these predicted miRNAs, 74 miRNA families were conserved in at least five mollusc species, and 53 miRNA families were conserved in half of the expected species, while miR-2, miR-29, miR-87, miR-184, and miRNA-1175 were identified in all of 35 mollusc species (). In fact, miR-467 genes were expanded in gastropod and cephalopod species, while miR-2 genes were strictly conserved and only found in invertebrates. In addition, let-7, miR-8, miR-10, miR-15, miR-17, and miR-181 were conserved in invertebrates and also expanded in vertebrates, including fish and mammals. Meanwhile, miR-750, miR-1175, miR-1986, miR-1990, miR-1991, miR-1993, miR-1994, and miR-20,001 were specifically observed in molluscs but not in other organisms. Two miRNAs with unconventional names, bantam and let-7, were also conserved in molluscs, except that let-7 was not found in A. dux via computational prediction and bantam was not found in nematodes and vertebrates. Furthermore, we found that miRNAs from the miR-2, miR-10, miR-25, miR-466, and miR-467 family were expanded from molluscs, with multiple or even dozens of transcript genes on the genome ().
Table 1. Genome-wide profiling of discovery of miRNAs in molluscs
Figure 1. Genome-wide scanning of miRNAs in various species of molluscs (A). The relationship between the number of scanned miRNAs and genome size. (B). The genomic miRNA density in molluscs. The density of miRNA is defined as the ratio of the number of predicted miRNAs and the genome size (Mb). (C). Conserved miRNA family in molluscs. The numbers in the box indicate the isoforms of the predicted miRNAs in each family. (D). PCA of molluscs and other organisms based on the predicted miRNA family. Arth: arthropod Drosophila melanogaster (Dme); Fish: Teleostei fish Danio rerio (Dre); Mamm: mammal Mus musculus (Mmu) and Homo sapiens (Hsa); Nema: nematode Caenorhabditis elegans (Cel); Biva: Bivalvia; Gast: Gastropoda; Ceph: Cephalopoda. E. Venn diagram of miRNA candidates using small-RNA sequencing and genome-wide scanning in Crassostrea gigas F. Venn diagram of miRNA candidates using small-RNA sequencing and genome-wide scanning in Pinctada fucata.
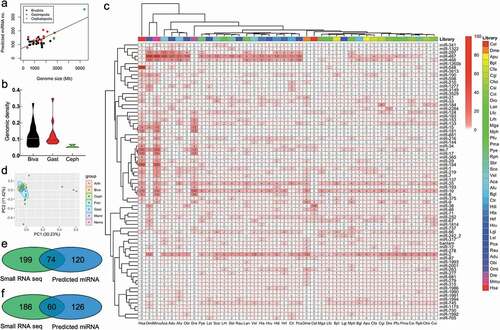
Based on the predicted miRNA family members, the bivalves and gastropods were clustered together and has a similar miRNA composition as cephalopods, arthropods, and nematodes, rather than fish or mammals by principal component analysis (PCA) (). The predicted miRNAs were also compared with the small RNA sequencing dataset of Crassostrea gigas [Citation25] and Pincatada fucata [Citation26]. Of those predicted miRNAs, 74 of 120 miRNAs (61.67%) were found in C. gigas’s small RNA sequencing-identified miRNAs (), and 69 of 126 (54.76%) miRNAs were found in P. fucata’s small RNA-verified miRNAs (). Eight miRNAs, namely let-7a, miR-279-3p, and miR-8-3p, which were abundant in multiple tissues, and miR-9a-3p, miR-183, miR-1986, miR-1990-3p, and miR-1993-3p, which were expressed in a tissue-specific manner based on small RNA sequencing in our previous study [Citation26], were chosen for miRNA authenticity verification in P. fucata. All of the miRNAs could be readily detected via stem-loop qRT-PCR, with various expression levels among tissues (Fig. S2). Specifically, miR-9a-3p, miR-1986, and miR-1990-3p were highly expressed in mantle tissues as compared with other tissues, which may indicate a vital role on the part of these miRNAs in the biomineralization process in P. fucata.
Genomic miRNA isoform and organization
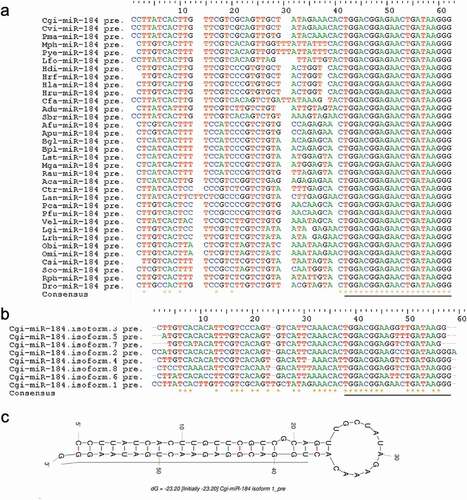
The miRNAs were generally annotated as a unique mature sequence, that is, the reference or canonical miRNA sequence. However, the recent development of deep sequencing has clearly shown that mature miRNAs can be heterogeneous in terms of length and/or sequence variants or isoforms in animals [Citation27]. Abundant miRNA isoforms, with variations in length and/or sequence, were also identified in the present study, yet some miRNAs were conserved among molluscs species. For instance, the mature sequence of miR-184 was conserved in all examined molluscs species, with a very similar precursor sequence (). Furthermore, we also identified a great deal of heterogeneous miRNA isoform variation in terms of length and/or sequence with respect to the corresponding canonical miRNAs in molluscs ().
Figure 2. Conserved miR-184 in molluscs (A). Alignment of precursors of miR-184 in molluscs. miR-184 shows conserved mature sequences in molluscs. (B). Alignment of precursors of C. gigas miR-184 isoforms. (C). Structure of precursor of Cgi-miR-184.isoform.1. The underline shows mature miRNA
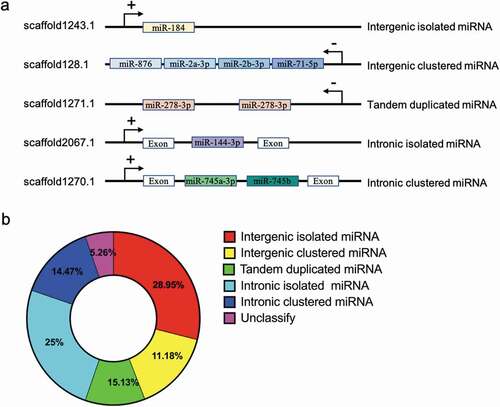
Clustered miRNAs are co-transcribed because they are generated from large primary transcripts within the genome [Citation28]. These clusters are often transcribed as a single unit, while some other clusters also emerged via the tandem duplication of a single miRNA [Citation29]. All predicted miRNAs had one or multiple mapping loci on the genome, with a recognizable precursor hairpin structure (). Some miRNA genes are located in cluster groups in intergenic and intronic protein coding genes (). Generally, intergenic miRNAs are transcribed from their own promoters, but intronic miRNAs are located in the same orientation as the host genes. In the case of miRNA genes on the genomic locus in P. fucata, as many as 62 (40.79%) miRNAs were distributed in clusters, and 82 (53.95%) precursors were intragenic, which suggests two main biogenetic sources of miRNAs ().
Figure 3. Genomic organization of miRNA genes in P. fucata (A). miRNA genes can be intergenic (alone or clustered) in the intron of non-coding RNA or protein coding genes (alone or clustered) or tandem duplicated on the genome. ‘+’ indicates miRNA genes transcribed from the sense strand, and ‘-’ indicates an antisense-strand assembly genome. (B). Composition of the various genomic organizations of miRNA genes in P. fucata. The majority of tandem duplicated miRNAs are located in intergenic region except for miR-29b located in intronic region
miRNA target prediction in molluscs
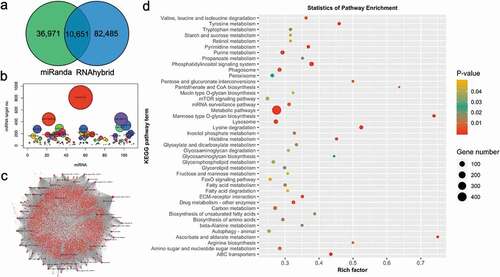
In order to have a deep understanding of miRNA function in molluscs, we performed target prediction using miRanda and RNAhybrid in C. gigas and P. fucata. Tens of thousands of target sites were anticipated to be targeted by miRNAs in P. fucata (). The genes predicted to bind with same miRNAs using two different methods were used for the subsequent functional analysis. A total of 7,278 genes were predicted to be targeted by miRNAs in P. fucata. Some miRNAs were predicted to regulate several or even hundreds of genes, and some genes were targeted by multiple miRNAs (). In P. fucata, miR-758 has the greatest number of predicted target genes, followed by miR-3897 and miR-10,280, while hundreds of the predicted genes were anticipated to be targeted by miR-2 family members (Table S2). The miRNAs perform post-transcriptional regulation in a complex network with genes (). The target prediction of miRNAs in C. gigas also shows similar characteristics (Fig. S3); miR-2 family members, such as miR-2a, miR-2b, and miR-2 c, have the greatest number of predicted target genes, followed by miR-3897 (Table S2). The predicted target genes were also submitted for Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathway annotation, and the outcomes showed a similarly enriched result in both of these two mollusc species. A total of 42 and 24 KEGG pathway terms, which were involved with glycol-metabolism, fatty acid metabolism, amino acid metabolism, and various signalling pathways, were enriched in P. fucata () and C. gigas (Fig. S3D), indicating that the miRNAs play various roles in physiological, biological, and molecular processes in molluscs.
Figure 4. Target prediction of miRNAs in P. fucata (A). Venn diagram of the number of predicted target sites between genes and genomic identified miRNAs using miRanda and RNAhybrid algorithms. (B). The number of target genes for each miRNA. (C). miRNAs-target genes regulatory network of genomic-predicted miRNAs in P. fucata. (D). Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathways, terms, and statistics of the KEGG pathway enrichment analysis for the predicted target genes of genomic miRNAs in P. fucata.
miRNA biogenesis pathway in molluscs
Based on homologous sequence searching, the key miRNA biogenesis factors were scanned in the mollusc species with complete sequences or long fragment sequences (). The amino acid sequences of miRNA biogenesis factors were listed in the supplementary sequences file. We reconstructed the structure of the key biogenesis factors, which contained the conserved domains for the biosynthesis, maturation, and transport of the miRNAs during the cellular process, as shown in P. fucata (). The phylogenetic analysis separated bivalves, gastropods, and cephalopods into three clear cluster groups in AGO2 (), DICER, and RAN, while gastropods and cephalopods are closer in terms of DGCR8, DROSHA, TARBP2, and XPO5 (Fig. S4).
Table 2. miRNA biogenesis factors identified in molluscs based on genome scanning
Figure 5. miRNA biogenesis factors and pathway in molluscs (A). Conserved domains of miRNA biogenesis factors in P. fucata. (B). Phylogenetic relationships of key miRNA biogenesis factor AGO2 in molluscs. Inferred protein sequences were aligned using ClustalX, and conserved positions were extracted using Gblocks and subjected to phylogenetic analysis. Homologous sequences from vertebrates (Vert), including Danio rerio (Dre), Mus musculus (Mmu), and Homo sapiens (Hsa), were used as outgroups. Biva is an abbreviation for Bivalvia, which is shaded in green; Gast for Gastropoda in yellow; and Ceph for Cephalopoda in blue. (C). A graphical reconstruction of miRNA biogenesis in molluscs. The miRNAs are transcribed by RNA polymerase II (Pol II) into primary miRNAs (pri-miRNA) and then processed by the DROSHA-DGCR8 complex into precursor miRNAs (pre-miR). The mature miRNA strand is highlighted in red. The pre-miR was transported from the nucleus into the cytoplasm by XPO5-RAN complex, where it was then loaded into the DICER-TARBP2 complex and then into an miRNA duplex and unwound. The mature strand is incorporated into the RNA-induced silencing complex (miRISC). Depending on an miRNA’s complementarity to a target mRNA, the miRISC-mediated gene regulation will occur via either translational repression or mRNA degradation
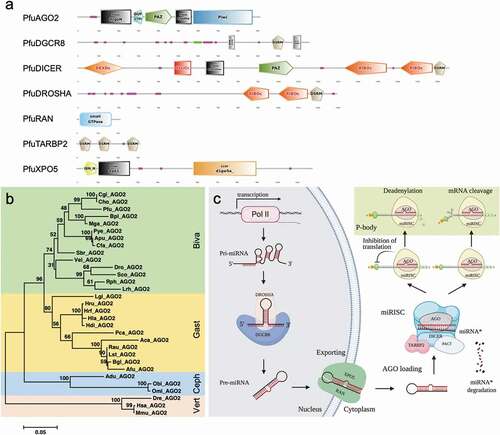
In animals, miRNAs are usually generated from various locations as clustered or independent transcriptional units on the genome. Based on previous knowledge, we summarized the miRNA biogenesis pathway in molluscs as shown in . The miRNA genes were transcribed from the transcriptional units by RNA polymerase II into a precursor transcript, namely primary miRNA (pri-miRNA). These pri-miRNAs have a hairpin structure that contains an imperfectly paired stem [Citation14]. In the nucleus, a microprocessor complex contains DROSHA, an RNase III endonuclease, and its cofactor DGCR8, which promote the pri-miRNAs’ adoption of a hairpin structure [Citation30]. DGCR8 binds the pri-miRNA stem and interacts with DROSHA to cleave the double-stranded stem and generate precursor miRNA (pre-miRNA) with a length of 60–80 nt [Citation31–33]. Then, the pre-miRNA was transferred into the cytoplasm and, subsequently, further processed by another RNase III enzyme, DICER, and its partner, TRBP2, for RNA binding [Citation34,Citation35]. The PAZ domain of DICER allows it to bind to the 3ʹ end of pre-miRNAs and thus cleave off the loop to generate a double-stranded miRNA duplex. The double-standed miRNA duplex is approximately 22 nt in length and associates with an AGO protein that rapidly unwinds this duplex; one strand, called the guide strand, is retained, whereas the other strand, the passenger or star (miRNA*) strand, is lost [Citation36]. After the unwinding of these duplexes by AGO, the passenger strand is degraded, whereas the guide strand (mature miRNA) binds to AGO and additional accessory proteins to form the microRNA-induced silencing complex (miRISC). Then, miRISC binds to mRNAs and inhibits gene expression, either by destabilizing the mRNA or by repression translation. The miRNA* is typically degraded by RISC, although some miRNAs* are thought to negatively regulate gene expression as mature miRNA. It is not known why some miRNAs* are functional. One hypothesis is that the strands could be used differently in response to extracellular or intracellular cues to regulate a more diverse set of protein-coding genes as needed, or strand selection could be tissue specific [Citation37]. Strand selection also limits regulation by miRNAs* generated from highly expressed precursors [Citation38].
Discussion
Over the last decade, a number of computational and experimental approaches have been approved to identify miRNAs in various animals and plants. Homology searching is the most accurate and useful method for miRNA identification within unknown species, and it relies on the conservation of miRNAs between closely related species [Citation39]. Here, we performed a genome-wide scanning of homologous miRNAs in molluscs. Hundreds of known miRNAs were identified in molluscs, and at least 50 family members are conserved in various molluscs. In addition to the conserved miRNAs in Metazoa, a number of molluscs-specific miRNAs were also identified. In addition, we also summarized the characteristics and functions of the predicted miRNAs in molluscs.
In the present study, the predicting miRNAs of C. gigas and P. fucata were also compared with small RNA sequencing results. Not all predicted miRNA were detected by sequencing in these two examples of molluscs, which may indicate that some miRNAs are specific to distinct developmental stages or tissues within an organism. The sequencing libraries of these two molluscs were constructed from a specific development stage or limited tissues [Citation25,Citation26]. This may explain the variation in miRNA candidates detected using small RNA sequencing and genome-wide prediction. These false-positive errors, however, cannot be avoided using predictive methods to identify miRNAs, regardless of how deep the sequencing is and which developmental stages are considered [Citation40]. Therefore, further experiments are needed to verify the existence of the miRNAs predicted in molluscs in the present study. Nevertheless, it is also worth predicting miRNAs in molluscs from an evolutionary standpoint, especially because molluscs are such an important, yet neglected, lineage of animals, and not much is known about their miRNAs.
Among these predicted miRNAs, five miRNAs, namely miR-2, miR-29, miR-87, miR-184, and miRNA-1175, were identified in all examined molluscs. In fact, miR-2 is an invertebrate-specific miRNA that plays a vital role in immune response and has many family members [Citation41]. Also, miR-184 was observed in all the examined molluscs, with a small number of isoforms, and it was evolutionarily conserved in all studied organisms, including Drosophila and vertebrates [Citation28]. In Drosophila, miR-184 was ubiquitously expressed in embryos, larvae, and adult tissues; its expression pattern shows dynamic changes during various developmental stages [Citation42]. In mammals, mature miR-184 is particularly enriched in the brain, testes, and corneal epithelium [Citation43]. Many targets for miR-184 have been verified, such as regulators of neurological development and apoptosis, and it has been suggested that miR-184 plays an essential role in development [Citation44]. Previous studies have also indicated that miR-184 is highly expressed in molluscs, such as P. martensii [Citation45], Tegillarca granosa [Citation46], and P. fucata [Citation26]. Bantam and let-7, with their unconventional miRNA names, were also conserved in molluscs. Bantam is an essential target of the Hippo signalling pathway and regulates cell proliferation, death, and survival in animals [Citation47]. Additionally, let-7, first discovered as a key developmental regulator in C. elegans, was one of the first two known miRNAs [Citation48] and has been implicated in the post-transcriptional control of innate immune responses to pathogenic agents [Citation49]. These miRNAs were conserved and had a long evolution history from Protozoa, suggesting that they may play crucial roles in essential biological processes in various organisms. However, a number of miRNAs, namely miR-750, miR-1175, miR-1986, miR-1990, miR-1991, miR-1993, miR-1994, and miR-2001, were specifically discovered in molluscs. The function of these miRNAs in biological development remains unclear in molluscs.
Initially, miRNA isoforms were considered to be sequencing errors, but more evidence has revealed that miRNA isoforms are actually miRNA variants that can have a high level of expression and biological activity [Citation50]. In fact, miRNA isoforms usually originate from imprecise cleavage by the DROSHA or DICER complex, which generates variants that show perfect sequence consistency with their pre-miRNA [Citation51]. Alternatively, miRNA isoforms could also be generated by post-transcriptional modifications due to enzymatic activity, which could either add or remove specific nucleotides at the 3′-end, while this would be rare at the 5ʹ-end [Citation52]. In the present study, we found that the miRNA isoforms may also be generated from different precursors on the genome. Although miRNA isoforms have been known for a long time, the functional significance of miRNA isoforms has remained elusive due to the limited research conducted. Therefore, the generation process and biological function of various miRNA isoforms will represent an important research topic in the near future.
The identification of miRNA targets is of central importance for a functional understanding of miRNA. We also performed a computational prediction of the targets of molluscs’ miRNAs using multiple bioinformatic tools. Thousands of targets were identified based on the physical sequence interaction with the RISC complex. Some miRNAs were predicted to bind to hundreds of target genes, and some targets were regulated by multiple miRNAs. The computational prediction of potential targets always yields a large proportion of false positives [Citation53,Citation54], but target validation failures could be due to erroneous target prediction, low-abundance targets, or miRNAs with limited or no activity because most of the target-miRNA interactions are biologically irrelevant; alternately, biological functionality may depend on the dose-sensitivity of the host gene [Citation55]. A complex miRNA-mediated regulatory network was predicted in molluscs, but it will require huge experimental work to confirm its existence. Experimental verification is vital for target prediction in the present study. To improve the understanding of miRNA function in molluscs, we further summarize the reported functional studies of mollusc miRNAs, most of which were detected based on genome-wide prediction.
Molluscs are representative animals that are widely distributed from freshwater to the deep sea. As successful settlers of the aquatic system, molluscs are remarkably resilient to harsh environmental conditions, including wide fluctuations in temperature, salinity, and hydraulic pressure, as well as prolonged air exposure and microbe-rich environments [Citation23]. Their extensive adaptive capabilities make molluscs interesting models to study. Recent advances in genomic studies of molluscs have provided insights into their immune and stress responses [Citation56–58]. Thousands of genes involved in immune and stress responses have been identified; however, a very limited number of miRNAs have been elucidated in molluscs. Thus far, only five miRNAs from Holiotis rufescens and 64 miRNAs from Lottia gigantean have been recorded by miRBase (Version 22.0). Recently, next-generational sequencing technology has been used to identify conserved and novel miRNAs in molluscs (). Additionally, the computational prediction of candidate miRNAs was performed using Patella vulgata [Citation59] and P. martensii [Citation60] genomic sequences. However, functional studies of specific miRNAs are still very rare in molluscs, especially those focusing on the biomineralization process ().
Table 3. Summary of identified miRNAs in molluscs
Table 4. Summary of validated miRNAs, their target genes, and functions in molluscs
In terms of biomineralization, molluscs are the most important organisms in marine ecosystems; however, only very limited studies have addressed the diversity and functional roles of miRNAs in such processes in molluscs. Among the miRNAs identified in molluscs in the present study, the expression of miR-29a [Citation61], miR-29b [Citation62], miR-125 [Citation63], miR-183 [Citation64], miR-200a [Citation65], miR-210 [Citation66], and miR-196a [Citation67] was reported by next-generation sequencing or RT-PCR methods. These miRNAs play an important role in the regulation of tooth and bone formation in vertebrates, scale mineralization in fish, shell or pearl formation in molluscs, and the formation of sea urchin spicules, suggesting crucial roles in the biomineralization process in organisms [Citation68–70]. To be more specific, miR-125b participated in the control of biomineralization during bone formation in animals [Citation71]. Also, miR-29a was involved in nacre formation and immune response by regulating neuropeptide Y receptor type 2 (Y2R) [Citation61], miR-2305 may participate in nacre formation by targeting pearlin in P. martensii [Citation68], and miR-4504 was involved in nacre colour formation in H. cumingii [Citation72]. These studies have greatly improved our understanding of miRNA function in biomineralization; however, the involvement of many other miRNAs in the biomineralization process still remains uninvestigated in molluscs.
Abundant miRNAs were also identified in mollusc haemocytes, which play a key role in the immune response as the main cellular effectors [Citation73–75]. An invertebrate-specific and immune-responsive miRNA, miR-2d, was proven to repress the synthesis and release of acetylcholine by targeting choline transporter-like protein (CTL1) and choline uptake in haemocytes during the early stage of pathogen infection and also proven to regulate nuclear factor kappa B2 protein (IκB2) expression and haemocyte phagocytosis during bacterial infection in the oyster C. gigas [Citation76,Citation77]. Similarly, miR-92d was also proven to indirectly regulate TNF expression by targeting lipopolysaccharide-induced TNF-α factor (LITAF3) [Citation78], while miR-29a was involved in the immune response by regulating neuropeptide Y receptor type 2 (Y2R) in pearl oyster P. martensii [Citation61]. Many conserved miRNAs identified in P. fucata, namely miR-92, miR-100, miR-124, miR125, miR-190, and let-7, also play a crucial role in immunity processes [Citation61,Citation79,Citation80]. These miRNAs were identified in most species, indicating that the miRNAs play a conventional role in immune response in molluscs.
The adaptation mechanism of molluscs to various environmental stressors encompasses specific molecular responses, and increasing evidence suggests that miRNA is one of the most important regulators in such molecular responses to various biotic and abiotic stressors. In previous studies, it was reported that under osmotic stress conditions, miR-92, miR-183, miR-184, miRNA-1984, and miRNA-2353, were differentially expressed in the gills of C. gigas, while miR-2353 and miR-3205 were differentially expressed in the gills of C. hongkongensis [Citation81]. Also, miR-183 was involved in the environmental stress response and functioned as a regulator of Taok1 to activate the mitogen-activated protein kinase pathway in response to stress and DNA damage caused by apoptosis [Citation82], and miR-365 was proven to contribute to host adaptation against desiccation stress by directly promoting the expression of HSP90AA1 in C. gigas [Citation83]. Hypoxia-inducible miRNAs are potentially involved in anti-apoptosis, regenerative mechanisms, and cell proliferation in squids Dosidicus gigas [Citation84]. In fact, miR-2001-5p was also up-regulated during hypoxia in squids, which has been proven to be associated with stress responses in shrimp, Rimicaris exoculata [Citation85], and eviscerated sea cucumbers, A. japonicus [Citation86]. Several miRNAs, including miR-1a, miR-34a, miR-29b, and miR-133, that are known to influence the cellular signalling pathway, energy metabolism, and apoptosis, were differentially expressed during extended periods of freezing and anoxia in Littorina littorea [Citation87]. Potentially, miR-2 and miR-153 may regulate the survival mechanism by targeting the Akt and Erk1/2 signalling pathways and myosin genes in milk snail, Otala lactea [Citation88]. Both miR-10 and miR-67 are potentially associated with gene regulation in the response to heavy metal Cd stress in T. granosa [Citation46]. Therefore, the occurrence of many of these miRNAs may play a critical role in the response to environmental stress in molluscs.
A few studies have also described the potential function of miRNAs in the growth and developmental process in molluscs. Specifically, miR-1990-3p, an identified mollusc-specific miRNA, showed a mantle-specific expression pattern in P. fucata [Citation26]. In C. gigas, another mollusc-specific miRNA, miR-1991-5p was found to be specifically expressed in adductor muscle [Citation25]. A previous study also reported that brain-specific miR-124, which is present in most organisms, responds to serotonin by derepressing CREB and enhancing serotonin-dependent long-term facilitation in Aplysia californica [Citation79]. Also, miR-124 was abundant in the adult pond snail’s, Lymnaea stagnalis’s, central nervous system (CNS), and its expression was up-regulated during both development and CNS regeneration, which indicate the conserved roles of this miRNA in neurogenesis [Citation89]. Many of these conserved miRNAs have also been identified in molluscs in the present study, indicating a conventional role on the part of miRNAs in various growth and developmental processes.
Conclusion
In the present study, we performed a genome-wide computational prediction of miRNAs in molluscs. Hundreds of conserved miRNAs were identified in these 35 molluscs. The miRNAs’ population, isoforms, and organization were characterized, as well as their potential function. The miRNA biogenesis pathway was complicated in that it recruited a number of factors, including AGO2, DGCR8, DICER, DROSHA, TRABP2, RAN, and XPO5. We also summarized the miRNAs’ function in biomineralization, immune and stress response, as well as growth and development in molluscs. Because miRNAs play a vital role in cellular and biological processes, this study will provide insight into miRNA biogenesis and functional understanding in molluscs, as well as other invertebrates.
Materials and methods
Genome-wide miRNA scanning
In order to identify newer miRNAs, direct cloning, next-generation sequencing, and computational techniques have been widely used in many species [Citation60,Citation90]. Here, we used the computational prediction of candidate miRNAs in various species of molluscs. The mature miRNAs of various animals were downloaded from miRBase (http://www.mirbase.org, released 22) and used for homologous sequence scanning in the genomes of molluscs. The genomic sequences of 35 species of molluscs were downloaded from a public database (). The homologous sequences of the animal miRNAs were identified using BLAST (Basic local alignment search tool), and the secondary structures of miRNA precursors were processed by miRDeep2 [Citation91]. Procedures for searching for candidate miRNAs were performed as described previously, with some modification. The following criteria were used to predict mature miRNAs and precursors: (1) predicted mature miRNAs were allowed to have only 0–4 mismatches in sequences with known mature miRNAs, (2) the mismatched nucleotides were not permitted in known miRNAs’ seed region (2–8 bp), and (3) miRNA precursors can fold into an appropriate hairpin secondary structure that contains mature miRNA sequences within one arm of the hairpin and has the smallest possible folding energy.
RNA extraction and quality control
Six adult P. fucata (sex ratio = 1:1) were collected from Mikimoto Pearl Research Institution Base at Mie Prefecture, Japan, in June 2019 and used for tissue collection. Adductor muscle, gill, mantle, and gonad tissue were collected and stored in 2 ml tubes with RNAlater (Thermo Fisher, Japan). The tissues samples were stored overnight at 4°C and then preserved at −80°C until use. Total RNA was isolated from the various tissues using an RNeasy Mini Kit (QIAGEN, Maryland, USA). An Agilent 2200 TapeStation (Agilent Technologies, Waldbronn, Germany) was used to determine the total RNA quantity and quality.
Validation of miRNAs by qRT-PCR
Eight predicted miRNAs were selected for miRNA validation in P. fucata using stem-loop qRT-PCR method [Citation92]. The designed primer sequences are listed in supplementary Table S3, and U6 snRNA was used as the internal reference for the stem-loop qRT-PCR. The protocols of RNA reverse transcription and stem-loop qRT-PCR were performed based on our previous study with P. fucata [Citation26]. The relative expression of each miRNA, quantified via stem-loop qRT-PCR, was presented as n = 6, while the Ct values were represented by the mean values of three independent replicates, and the relative expression levels were calculated using the 2−∆∆Ct method [Citation93].
The miRNA target prediction
The 3′UTRs sequences were isolated from the C. gigas and P. fucata gene-annotated results and used for miRNA-target prediction in these two molluscs. Target predictions for miRNAs were performed by RNAhybrid [Citation94] and miRanda [Citation95]. For the RNAhybrid algorithm, free energy (e) and p-value (p) were used to estimate the interaction between miRNAs and mRNAs. The conditions for functional targets were e < −20 kcal/mol and p < 0.05, a score > 100, and a free energy < −10 kcal/mol in the miRanda algorithm. The genes that were simultaneously predicted to be targeted with the same miRNAs using two different algorithms were considered as target genes of special miRNA. The predicted target genes were submitted to KOBAS 3.0 (http://kobas.cbi.pku.edu.cn/kobas3) for KEGG pathway annotation [Citation96]. The network of miRNAs and targeted genes was drawn using NetworkAnalyst [Citation97].
The miRNA biogenesis pathway
Sequence coding for miRNA biogenesis factors, including AGO2, DGCR8, DICER, DROSHA, TRABP2, RAN, and XPO5, has been methodically performed in the genomes of A. californica, Biomphalaria glabrata, Chlamys farreri, C. gigas, Crassostrea virginica, L. gigantea, Mytilus galloprovincialis, Patinopecten yessoensis, Octopus bimaculoides, Pomacea canaliculate, and P. fucata, as well as the transcriptomes of other molluscs. The sequences of these verified miRNA biogenesis factors were used for homologous sequence searching in genomic sequences using exonerate V2.2.0 [Citation98], and the protein domain organization of the key factors of miRNA biogenesis was constructed using SMART [Citation99]. The inferred protein sequences were aligned using ClustalX, Version 2.1 [Citation100]. Subsequently, the fasta alignments were analysed using Gblocks V.0.91 [Citation101] to extract conserved positions (positions common to 51% of the locally aligned sequences). Trees were built using neighbour-joining clustering methods, with 1,000 bootstrap replicates, using MEGA Version 6.06 [Citation102].
Author contributions
AS, WS, and KS designed the study. HS and YK performed all bioinformatics analyses. HS performed the RT-PCR analysis. AS and HS wrote the manuscript. AM collected the datasets, with assistance from KS, and helped to draft the manuscript. All authors read and approved the final manuscript.
Supplemental Material
Download Zip (4.2 MB)Disclosure statement
The authors declare no competing interest.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Kim N. Small RNAs: classification, biogenesis, and function. Mol Cells. 2005;19(1):1–15.
- Kawamura Y, Saito K, Kin T, et al. Drosophila endogenous small RNAs bind to Argonaute 2 in somatic cells. Nature. 2008;453(7196):793–797.
- Papenfort K, Vogel J. Multiple target regulation by small noncoding RNAs rewires gene expression at the post-transcriptional level. Res Microbiol. 2009;160(4):278–287.
- Carthew R, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell. 2009;136(4):642–655.
- Kim V, Han J, Siomi M. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. 2009a;10(2):126–139.
- Carninci P. Molecular biology: the long and short of RNAs. Nature. 2009;457(7232):974–975.
- Kim S, Song M, Min H, et al. miRNA biogenesis-associated RNase III nucleases Drosha and Dicer are upregulated in colorectal adenocarcinoma. Oncol Lett. 2017;14(4):4379–4383.
- Lee R, Feinbaum R, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75(5):843–854.
- Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75(5):855–862.
- Kozomara A, Birgaoanu M, Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2019;47(D1):D155–62.
- Ibanez-Ventoso C, Vora M, Driscoll M. Sequence relationships among C. elegans, D. melanogaster and human microRNAs highlight the extensive conservation of microRNAs in biology. PLoS One. 2008;3(7):e2818.
- Sperling E, Vinther J, Moy V, et al. MicroRNAs resolve an apparent conflict between annelid systematics and their fossil record. Proc R Soc B. 2009;276(1677):4315–4322.
- Wheeler B, Heimberg A, Moy V, et al. The deep evolution of metazoan microRNAs. Evol Dev. 2009;11(1):50–68.
- Bartel D. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297.
- Urbich C, Kuehbacher A, Dimmeler S. Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc Res. 2008;79(4):581–588.
- Di L, Calin G, Croce C. MicroRNAs: fundamental facts and involvement in human diseases. Birth Defects Res C. 2006;78(2):180–189.
- Bushati N, Cohen S. MicroRNA functions. Annu Rev Cell Dev Biol. 2007;23(1):175–205.
- Vasudevan S, Tong Y, Steitz J. Switching from repression to activation: microRNAs can up-regulate translation. Science. 2007;318(5858):1931–1934.
- Mortensen R, Serra M, Steitz J, et al. Posttranscriptional activation of gene expression in Xenopus laevis oocytes by microRNA-protein complexes (microRNPs). Proc Natl Acad Sci USA. 2011;108(20):8281–8286.
- Place R, Li L, Pookot D, et al. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc Natl Acad Sci USA. 2008;105(5):1608–1613.
- Weinmann L, Hock J, Ivacevic T, et al. Importin 8 is a gene silencing factor that targets argonaute proteins to distinct mRNAs. Cell. 2009;136(3):496–507.
- Younger S, Corey D. Transcriptional gene silencing in mammalian cells by miRNA mimics that target gene promoters. Nucleic Acids Res. 2011;39(13):5682–5691.
- Guo X, He Y, Zhang L, et al. Immune and stress responses in oysters with insights on adaptation. Fish Shellfish Immunol. 2015;46(1):107–119.
- Green T, Raftos D, Speck P, et al. Antiviral immunity in marine molluscs. J Gen Virol. 2015;96(9):2471–2482.
- Xu F, Wang X, Feng Y, et al. Identification of conserved and novel microRNAs in the Pacific oyster Crassostrea gigas by deep sequencing. PLoS One. 2014;9(8):e104371.
- Huang S, Ichikawa Y, Yoshitake K, et al. Identification and characterization of micrornas and their predicted functions in biomineralization in the pearl oyster (Pinctada fucata). Biology (Basel). 2019;8(2):47.
- Neilsen C, Goodall G, Bracken C. IsomiRs-the overlooked repertoire in the dynamic microRNAome. Trends Genet. 2012;28(11):544–549.
- Aboobaker A, Tomancak P, Patel N, et al. Drosophila microRNAs exhibit diverse spatial expression patterns during embryonic development. Proc Natl Acad Sci USA. 2005;102(50):18017–18022.
- Marco A, Ninova M, Ronshaugen M, et al. Clusters of microRNAs emerge by new hairpins in existing transcripts. Nucleic Acids Res. 2013;41(16):7745–7752.
- Rosani U, Pallavicini A, Venier P. The miRNA biogenesis in marine bivalves. PeerJ. 2016;4:e1763.
- Lee Y, Ahn C, Han J, et al. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425(6956):415–419.
- Denli A, Tops B, Plasterk R, et al. Processing of primary microRNAs by the microprocessor complex. Nature. 2004;432(7014):231–235.
- Han J, Lee Y, Yeom K, et al. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004;18(24):3016–3027.
- Ketting R, Fischer S, Bernstein E, et al. Dicer functions in RNA interferen and in synthesis of small RNA involved in developmental timing in C. elegans. Ele Genes Dev. 2001;15(20):2654–2659.
- Hutvagner G, McLachlan J, Pasquinelli A, et al. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science. 2001;293(5531):834–838.
- Kawamata T, Seitz H, Tomari Y. Structural determinants of miRNAs for RISC loading and slicer independent unwinding. Nat Struct Mol Biol. 2009;16(9):953–960.
- Ro S, Park C, Young D, et al. Tissue-dependent paired expression of miRNAs. Nucleic Acids Res. 2007;35(17):5944–5953.
- Hu HY, Yan Z, Xu Y, et al. Sequence features associated with microRNA strand selection in humans and flies. BMC Genomics. 2009;10(1):413.
- Yones C, Stegmayer G, Milone DH. Genome-wide pre-miRNA discovery from few labeled examples. Bioinformatics. 2018;34(4):541–549.
- Prochnik SE, Rokhsar DS, Aboobaker AA. Evidence for a microRNA expansion in the bilaterian ancestor. Dev Genes Evol. 2007;217(1):73–77.
- Marco A, Hooks K, Griffiths-Jones S. Evolution and function of the extended miR-2 microRNA family. RNA Biol. 2012;9(3):242–248.
- Iovino N, Pane A, Gaul U. miR-184 has multiple roles in Drosophila female germline development. Dev Cell. 2009;17(1):123–133.
- Nomura T, Kimura M, Horii T, et al. MeCP2-dependent repression of an imprinted miR-184 released by depolarization. Hum Mol Genet. 2008;17(8):1192–1199.
- Li P, Peng J, Hu J, et al. Localized expression pattern of miR-184 in Drosophila. Mol Biol Rep. 2011;38(1):355–358.
- Jiao Y, Zheng Z, Du X, et al. Identification and characterization of microRNAs in pearl oyster Pinctada martensii by Solexa deep sequencing. Mar Biotechnol. 2013;16(1):54–62.
- Bao Y, Zhang L, Dong Y, et al. Identification and comparative analysis of the Tegillarca granosa haemocytes microRNA transcriptome in response to Cd using a deep sequencing approach. PLoS One. 2014;9(4):e93619.
- Nolo R, Morrison CM, Tao C, et al. The bantam microRNA is a target of the hippo tumor-suppressor pathway. Curr Biol. 2006;16(19):1895–1904.
- Reinhart B, Slack F, Basson M, et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403(6772):901–906.
- Schulte L, Eulalio A, Mollenkopf H, et al. Analysis of the host microRNA response to Salmonella uncovers the control of major cytokines by the let-7family. Embo J. 2011;30(10):1977–1989.
- Muller H, Marzi M, Nicassio F. IsomiRage: from functional classification to differential expression of miRNA isoforms. Front Bioeng Biotech. 2014;2:38.
- Starega-Roslan J, Witkos T, Galka-Marciniak P, et al. Sequence features of Drosha and Dicer cleavage sites affect the complexity of isomiRs. Int J Mol Sci. 2015;16(4):8110–8127.
- Newman M, Mani V, Hammond S. Deep sequencing of microRNA precursors reveals extensive 3ʹ end modification. RNA. 2011;17(10):1795–1803.
- Mockly S, Seitz H. Inconsistencies and limitations of current MicroRNA target identification methods. Methods Mol Biol. 2019;1970:291–314.
- Fridrich A, Hazan Y, Moran Y. Too many false targets for microRNAs: challenges and pitfalls in prediction of miRNA targets and their Gene Ontology in model and non-model organisms. Bioessays. 2019;41(4):e1800169.
- Pinzón N, Li B, Martinez L, et al. microRNA target prediction programs predict many false positives. Genome Res. 2017;27(2):234–245.
- Li Y, Sun X, Hu X, et al. Scallop genome reveals molecular adaptations to semi-sessile life and neurotoxins. Nat Commun. 2017;8(1):1721.
- Sun J, Zhang Y, Xu T, et al. Adaptation to deep-sea chemosynthetic environments as revealed by mussel genomes. Nat Ecol Evol. 2017;1(5):121.
- Liu C, Zhang Y, Ren Y, et al. The genome of the golden apple snail Pomacea canaliculata provides insight into stress tolerance and invasive adaptation. GigaScience. 2018;7(9):giy101.
- Kenny N, Namigai E, Marletaz F, et al. Draft genome assemblies and predicted microRNA complements of the intertidal lophotrochozoans Patella vulgata (Mollusca, Patellogastropoda) and Spirobranchus (Pomatoceros) lamarcki (Annelida, Serpulida). Mar Genom. 2015;24:139–146.
- Zheng Z, Jiao Y, Du X, et al. Computational prediction of candidate miRNAs and their potential functions in biomineralization in pearl oyster Pinctada martensii. Saudi J Biol Sci. 2016;23(3):372–378.
- Tian R, Zheng Z, Huang R, et al. miR-29a participated in nacre formation and immune response by targeting Y2R in Pinctada martensii. Int J Mol Sci. 2015;16(12):29436–29445.
- Li Z, Hassan M, Jafferji M, et al. Biological functions of miR-29b contribute to positive regulation of osteoblast differentiation. J Biol Chem. 2009;284(23):15676–15684.
- Mizuno Y, Yagi K, Tokuzawa Y, et al. miR-125b inhibits osteoblastic differentiation by down-regulation of cell proliferation. Biochem Biophys Res Commun. 2008;368(2):267–272.
- Zheng Z, Du X, Xiong X, et al. PmRunt regulated by Pm-miR-183 participates in nacre formation possibly through promoting the expression of collagen VI-like and Nacrein in pearl oyster Pinctada martensii. PLoS One. 2017;12(6):e0178561.
- Itoh T, Nozawa Y, Akao Y. MicroRNA-141 and −200a are involved in bone morphogenetic protein-2-induced mouse pre-osteoblast differentiation by targeting distal-less homeobox 5. J Biol Chem. 2009;284(29):19272–19279.
- Mizuno Y, Tokuzawa Y, Ninomiya Y, et al. miR-210 promotes osteoblastic differentiation through inhibition of AcvR1b. FEBS Lett. 2009;583(13):2263–2268.
- Kim Y, Bae S, Yu S, et al. miR-196a regulates proliferation and osteogenic differentiation in mesenchymal stem cells derived from human adipose tissue. J Bone Miner Res. 2009b;24(5):816–825.
- Jiao Y, Zheng Z, Tian R, et al. MicroRNA, Pm-miR-2305, participates in nacre formation by targeting pearlin in pearl oyster Pinctada martensii. Int J Mol Sci. 2015;16(9):21442–21453.
- Stepicheva NA, Song J. MicroRNA-31 modulates skeletal patterning in the sea urchin embryos. Development. 2015;142(21):3769–3780.
- Li S, Chen W, Zhan A, et al. Identification and characterization of microRNAs involved in scale biomineralization in the naked carp Gymnocypris przewalskii. Comp Biochem Physiol Part D. 2018;28:196–203.
- Xia Z, Chen C, Chen P, et al. MicroRNAs and their roles in osteoclast differentiation. Front Med. 2011;5(4):414–419.
- Chen X, Zhang M, Zhang J, et al. miR-4504 is involved in nacre color formation in Hyriopsis cumingii. Biochem Biophys Res Commun. 2019a;517(2):210–215.
- Martin-Gomez L, Villalba A, Kerkhoven RH, et al. Role of microRNAs in the immunity process of the flat oyster Ostrea edulis against bonamiosis. Infect Genet Evol. 2014;27:40–50.
- Zhou Z, Wang L, Song L, et al. The identification and characteristics of immune-related microRNAs in haemocytes of oyster Crassostrea gigas. PLoS One. 2014;9(2):e88397.
- Burgos-Aceves M, Cohen A, Smith Y, et al. A potential microRNA regulation of immune-related genes in invertebrate haemocytes. Sci Total Environ. 2018;621:302–307.
- Chen H, Zhou Z, Wang H, et al. An invertebrate-specific and immune-responsive microRNA augments oyster haemocyte phagocytosis by targeting CgIκB2. Sci Rep. 2016a;6(1):29591.
- Chen H, Zhou Z, Wang L, et al. An invertebrate-specific miRNA targeted the ancient cholinergic neuroendocrine system of oyster. Open Biol. 2016b;6(8):160059.
- Chen H, Jiang S, Wang L, et al. Cgi-miR-92d indirectly regulates TNF expression by targeting CDS region of lipopolysaccharide-induced TNF-α factor 3 (CgLITAF3) in oyster Crassostrea gigas. Fish Shellfish Immunol. 2016c;55:577–584.
- Rajasethupathy P, Fiumara F, Sheridan R, et al. Characterization of small RNAs in Aplysia reveals a role for miR-124 in constraining synaptic plasticity through CREB. Neuron. 2009;63(6):803–817.
- Li Z, Wu F, Brant S, et al. IL-23 receptor regulation by Let-7f in human CD4+Memory T cells. J Immunol. 2011;186(11):6182–6190.
- Zhao X, Yu H, Kong L, et al. High throughput sequencing of small RNAs transcriptomes in two Crassostrea oysters identifies microRNAs involved in osmotic stress response. Sci Rep. 2016;6(1):22687.
- Patel M, Cai Q, Ding D, et al. The miR-183/Taok1 target pair is implicated in cochlear responses to acoustic trauma. PloS One. 2013;8(3):e58471.
- Chen H, Xin L, Song X, et al. A norepinephrine-responsive miRNA directly promotes CgHSP90AA1 expression in oyster haemocytes during desiccation. Fish Shellfish Immunol. 2017;64:297–307.
- Hadj-Moussa H, Logan S, Seibel B, et al. Potential role for microRNA in regulating hypoxia-induced metabolic suppression in jumbo squids. BBA Gene Regul Mechs. 2018;1861(6):586–593.
- Zhou Y, He Y, Wang C, et al. Characterization of miRNAs from hydrothermal vent shrimp Rimicaris exoculate. Mar Genom. 2015;24:371–378.
- Wang Q, Yang H, Li X, et al. Understanding regulation of microRNAs on intestine regeneration in the sea cucumber Apostichopus japonicus using high- throughput sequencing. Comp Biochem Physiol D. 2017;22:1–9.
- Biggar K, Kornfeld S, Maistrovski Y, et al. MicroRNA regulation in extreme environments: differential expression of microRNAs in the intertidal snail Littorina littorea during extended periods of freezing and anoxia. Genomics, Proteomics & Bioinformatics. 2012;10(5):302–309.
- Hoyeck M, Hadj-Moussa H, Storey K. Estivation-responsive microRNAs in a hypometabolic terrestrial snail. PeerJ. 2019;7:e6515.
- Walker S, Spencer GE, Necakpv A, et al. Identification and characterization of microRNAs during retinoic acid-induced regeneration of a molluscan central nervous system. Int J Mol Sci. 2018;19(9):2741.
- Wang X, Zhang J, Li F, et al. MicroRNA identification based on sequence and structure alignment. Bioinformatics. 2005;21(18):3610–3614.
- Friedlander M, Mackowiak S, Li N, et al. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012;40(1):37–52.
- Chen C, Ridzon D, Broomer A, et al. Real-time quantification of micro-RNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005;33(20):e179.
- Pfaffl M. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2011;29(9):e45.
- Rehmsmeier M, Steffen P, Hochsmann M, et al. Fast and effective prediction of microRNA/target duplexes. RNA. 2014;10(10):1507–1517.
- Enright A, John B, Gaul U, et al. MicroRNA targets in Drosophila. Genome Biol. 2003;5(1):R1.
- Xie C, Mao X, Huang J, et al. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011;39(suppl_2):W316–22.
- Zhou G, Soufan O, Ewald J, et al. NetworkAnalyst 3.0: a visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acid Res. 2019;47(W1):W234–41.
- Slater G, Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6(1):31.
- Letunic I, Bork P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2017;46(D1):493–496.
- Larkin M, Blackshields G, Brown N, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–2948.
- Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17(4):540–552.
- Tamura K, Stecher G, Peterson D, et al. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–2729.
- Chen G, Zhang C, Jiang F, et al. Bioinformatics analysis of hemocyte miRNAs of scallop Chlamys farreri against acute viral necrobiotic virus (AVNV). Fish Shellfish Immunol. 2014;37(1):75–86.
- Chen H, Wang L, Zhou Z, et al. The comprehensive immunomodulation of NeurimmiRs in haemocytes of oyster Crassostrea gigas after acetylcholine and norepinephrine stimulation. BMC Genomics. 2015;16(1):942.
- Wei P, He P, Zhang X, et al. Identification and characterization of microRNAs in the gonads of Crassostrea hongkongensis using high-throughput sequencing. Comp Biochem Physi D. 2019;31:100606.
- Chen X, Bai ZY, Li JL. The mantle exosome and microRNAs of Hyriopsis cumingii involved in nacre color formation. Mar Biotechnol. 2019b;21(5):634–642.
- Yu D, Wu H, Peng X, et al. Profiling of microRNAs and mRNAs in marine mussel Mytilus galloprovincialis. Comp Biochem Phys C. 2020;230:108697.
- Jiao Y, Zheng Z, Du X, et al. Identification and Characterization of MicroRNAs in Pearl Oyster Pinctada martensii by Solexa Deep Sequencing. Mar Biotechnol. 2014;16(1):54–62.
- Picone B, Rhode C, Roodt-wilding R. Identification and characterization of miRNAs transcriptome in the South African abalone, Haliotis midae. Mar Genom. 2017;31:9–12.
- Zhu X, Chen Y, Zhang Z, et al. A species-specific miRNA participates in biomineralization by targeting CDs regions of Prisilkin-39 and ACCBP in Pinctada fucata. Sci Rep. 2020;10(1):8971.