
ABSTRACT
Versatile RNA modifications play important roles in post-transcriptional regulations of gene expression, among which glycosylation modifications on small RNAs emerge as a novel clade whose characteristics need further interrogations. Here, we demonstrated that the sequence pattern around RNA glycosylation sites was not random and could be exploited for glycosylation site prediction. A machine learning predictor, GlyinsRNA, which integrated multiple RNA sequence representation encodings, was established. GlyinsRNA achieved AUROC (area under the receiver operating characteristic curve) of 0.7933 and 0.7979 in five-fold cross-validation and independent tests, respectively. GlyinsRNA was implemented as an online webserver, where both the predicted glycosylation sites and the overrepresented RNA-binding protein (RBP)-related motifs were annotated to facilitate the users. GlyinsRNA webserver is freely available at http://www.rnanut.net/glyinsrna.
Introduction
Various RNA modifications are extensively involved in the post-transcriptional gene expression regulation processes like RNA localization, RNA degradation, alternative splicing and translational efficiency regulation [Citation1]. Although more than one hundred types of RNA modification had been discovered [Citation1], just recently, the glycosylation modification has been shown to further extend the scope of RNA modification world [Citation2]. Unlike most of the known types of RNA modifications, in glycosylation, instead of typical small chemical group (e.g. methyl- or acetyl-group), a much larger carbohydrate residue (e.g. sialic acid and fucose) was conjugated onto the RNA substrates. In the leading reports from Flynn et al., RNA glycosylation was mainly enriched on small RNAs and plays roles on RNA localization and RNA recognition, suggesting its functional importance in cell signalling [Citation2]. However, the mechanism-of-action for RNA glycosylation remains to be fully elucidated, of which whether and how the sequence pattern contributes to this process has not been investigated.
In this study, we demonstrated that the sequence context around RNA glycosylation sites contains predictive information for RNA glycosylation, where both the positional sequence pattern and the RNA-binding protein (RBP)-related motifs are distributed in a non-random fashion. Based on these observations, GlyinsRNA, a sequence-based machine learning predictor of glycosylation sites on small RNA, was established.
Results and discussion
Non-random sequence pattern around RNA glycosylation sites
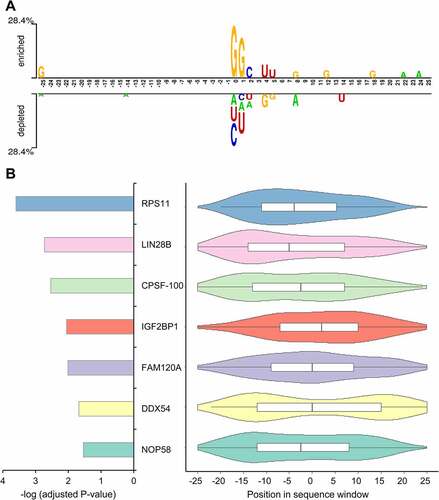
The sequence context around one site can be represented by a sequence window of 51 nucleotides (51 nt) with the modification sites at the centre. shows the positional sequence pattern of RNA glycosylation sites, where several positions, especially those proximal to the central glycosylation site (e.g. positions 0, +1, +2, +4 and +5), show non-random nucleotide propensities. On the other hand, however, no positional nucleotide propensity is observed for most of the other positions. One possible explanation is that there are position-independent sequence patterns (like short motifs) in addition to the observed positional sequence patterns. To test this possibility, we scanned the sequence windows with the pre-computed binding motifs of known RBPs. Indeed, the motifs of seven RBPs (RPS11, LIN28B, CPSF-100, IGF2BP1, FAM120A, DDX54 and NOP58) were significantly overrepresented in the sequence windows of the glycosylation sites in comparison with those of the non-modified sites (, bar plots), and most of the results could be still observed with shorter window sizes until those of 37 nt (Supplementary Fig. S1), indicating plausible functional links between glycosylated RNA and these RBP regulators. Weak or moderate position preferences were observed when checking the positional distribution of few overrepresented motifs along the sequence windows (, violin plots). For example, RPS11 and LIN28B have a tendency of binding to the upstream regions of glycosylation sites. Nonetheless, no strong position-biased distributions could be observed for any of these motifs. These results suggest the largely position-independent, short motif-like sequence patterns around glycosylation sites.
Figure 1. The positional and position-independent sequence pattern around the RNA glycosylation sites. (a) The positional sequence pattern around glycosylation sites. (b) The overrepresented RBP-related motifs found in the flanking sequence windows of glycosylation sites
Sequence-based prediction of RNA glycosylation site by machine learning models
Based on the above observations, we employed multiple sequence encoding schema to depict this non-random sequence pattern and trained random forest models based on these sequence feature encodings. We firstly optimized the window size k for each encoding. The performances of prediction models changing with the window size were summarized in Supplementary Fig. S2. The optimal window sizes of models by six encodings (PSNSP, PSDSP, One-hot, KNF, KSNPF, and PseKNC) were 51 nt, 51 nt, 51 nt, 17 nt, 19 nt and 37 nt, respectively.
Then, we constructed the models using the optimal window size for each encoding. As shown in , the best-performed model in cross-validation is that based on the PSDSP encoding (AUROC = 0.7704). We further tested if combining multiple models could achieve better performance. As a result, a predictor combining three encodings (PSDSP, One-hot and KNF) was found to be the most competitive one (AUROC = 0.7933). Interestingly, One-hot [Citation3,Citation4] and KNF [Citation5,Citation6] encodings were designed to represent positional and position-independent sequence patterns, while PSDSP [Citation7] encoding considered both aspects. Thus, the selection of the best encoding combination again suggests that both the positional and the position-independent sequence patterns provide predictive information for glycosylation sites.
Figure 2. Predictor performance evaluation results. (a) The ROC curve showing the performance of single and combined random forest models in five-fold cross-validation. (b) The ROC curve showing the performance of predictor on independent test dataset
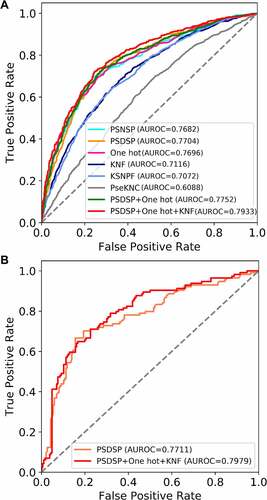
To further validate the predictor performance, we tested the combined predictor on the independent test dataset. Samples in this dataset were not used for model training nor for model parameter and model combination selection and therefore could serve as a more rigorous and unbiased validation of the performance. On the independent test dataset (), the combined predictor (AUROC = 0.7979) also outperformed the best single-encoding-based model (AUROC = 0.7711). We also tried to evaluate the prediction model performance by using the precision–recall curve and found similar tendency (Supplementary Fig. S3).
GlyinsRNA: webserver for small RNA glycosylation site prediction
Based on the above performance evaluation results, we finally selected the combined predictor to build our RNA glycosylation site prediction tool, i.e. GlyinsRNA. GlyinsRNA has been implemented as an online webserver, which requires only RNA sequences as its input. Users could also select one stringent threshold according to their expected specificity level. Using a low-specificity threshold means to report more low-scored prediction results and therefore allow a higher tolerance to false-positive results. Here, we provided six alternatives for specificity level to users, including 99.9%, 99.5%, 99.0%, 95.0%, 90.0% and 85.0%. The primary output of the webserver is the position and score of each predicted glycosylation site. Besides, the sequence context around each predicted glycosylation site is also displayed, where both the glycosylation sites and overrepresented RBP-related motifs (if any) are highlighted by the distinct colours (Supplementary Fig. S4). We hope this easy-to-use webserver would facilitate further experimental investigations of small RNA glycosylation in the future.
Materials and methods
Dataset
The genomic coordinate annotation of glycosylation site was obtained from GEO (accession: GSE136967). The sites exclusively observed on the glycoRNA library (i.e. RNA library with glycosylation modification) were selected as the positive samples, while those only observed in the input library (i.e. background RNA library) but not in the glycoRNA library were assigned as the unmodified negative samples. Compared to the random negative sites, such selection could exclude unrelated sites and therefore is more rigorous. We primarily obtained 2720 positive and 5078 negative sites, which were randomly separated into training dataset and independent test dataset with the proportion of 7:3 (which is same for both the positive samples and the negative samples) to construct and assess the predictor. After mapping the sample sites to small RNA sequences, we employed CD-HIT-EST tool [Citation8] to remove the redundant sequence samples with 90% identity threshold, by which 857 positive samples and 1068 negative samples were retained. Among all retained sample sequences, 114 positive and 186 negative samples were in the independent test set, while the rest were in the training set (with no overlap between the training and the testing samples).
Sequence pattern analysis
The sequence context around one site can be represented by a sequence window of k nucleotides with the modification/unmodified sites at the centre. The window size k was preliminarily optimized as 51 nt according to the cross-validation performance of the best-performed encodings. Meanwhile, we also explored the enrichment of RBP motifs with other window sizes and observed overrepresentation of most of the seven RBP motifs in shorter windows until those of 37 nt (Supplementary Fig. S1). If a sequence window extends beyond the border of an RNA, gap characteristics were filled to ensure all sequence windows were aligned to the centre. To investigate the positional sequence pattern of glycosylation sites, the aligned positive and negative sequence windows were analysed by TwoSampleLogo server [Citation9]. Besides, we screened the significantly overrepresented RBP motifs in the positive sequence windows (in contrast to the negative sequence windows) by the AME tool of MEME Suite [Citation10]. The Bonferroni adjusted P-value <0.05 was adopted as the significance threshold for the motif overrepresentation. The known RBP motifs were derived from the high-throughput CLIP-Seq experiment datasets, with the detailed description available in the previous work [Citation11].
Predictor construction and evaluation
Here, the One-hot [Citation3,Citation4], PSNSP [Citation12], PSDSP [Citation7], KNF [Citation5,Citation6], KSPNF [Citation4,Citation13] and PseKNC [Citation14] encodings were employed to depict the sequence pattern around glycosylation sites. These encodings are briefly introduced as following:
1) One-hot encoding translates four nucleotides, A, C, G, T, into binary numeric vectors, 0001,0010,0100,1000.
2) Position-specific nucleotide sequence profile (PSNSP) encoding displays the difference in frequencies of four nucleotides between positive samples and negative samples at each position of the sequence window.
3) Position-specific dinucleotide sequence profile (PSDSP) encoding is similar to PSNSP encoding, while it compares the frequency difference in dinucleotide between positive samples and negative samples.
4) K-nucleotide frequencies (KNF) encoding depicts the frequencies of continuous k-nucleotide on the sequence. The selection of K was preliminarily optimized as 2, 3 and 4.
5) K-spaced nucleotide pair frequencies (KSPNF) encoding also describes the frequencies of dinucleotide on the sequence. The dinucleotide is split by K-random nucleotides, and selection of K was preliminarily optimized as 0, 1, 2, 3 and 4.
6) Pseudo k-tuple nucleotide composition (PseKNC) encoding is the combination of the frequency and the physico-chemical property of dinucleotide or trinucleotide.
For each encoding, a random forest model was trained, and the prediction results from each were combined by the weighted summing schema where the weights of all models sum to 1. To determine the optimal window sizes of sequences, we trained models with various windows from 13 nt (larger than the RBP motif length which is 12 nt) to 51 nt with the step of 2 and selected optimal window sizes for each encoding. The best combination of random forest models was selected based on five-fold cross-validation performance. Besides, we also tested the models on the independent test set to avoid overestimation of performance. The performance was measured by typical area under the receiver operating characteristic curve (AUROC) metric. Finally, the predictor was implemented as an online webserver which was constructed under the ‘Linux + Apache + PHP’ framework.
Author contributions
CC established and tested the models. CC constructed the online webserver. CC and XW performed the sequence analysis. YZ and CC prepared and revised the manuscript. YZ supervised the study.
Supplemental Material
Download MS Word (1.6 MB)Acknowledgments
We thank Prof. Qinghua Cui, Yiran Zhou and Wanqing Zhao for their suggestions.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The RNA glycosylation site data from Flynn et al. is publicly available in public GEO series (accession: GSE136967) https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE136967here
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Wiener D, Schwartz S. The epitranscriptome beyond m(6)A. Nat Rev Genet. 2021;22(2):119–131.
- Flynn RA, Pedram K, Malaker SA, et al. Small RNAs are modified with N-glycans and displayed on the surface of living cells. Cell. 2021;184(12):3109–24 e22.
- Uriarte-Arcia AV, Lopez-Yanez I, Yanez-Marquez C. One-hot vector hybrid associative classifier for medical data classification. PloS One. 2014;9(4):e95715.
- Zhou Y, Zeng P, Li YH, et al. SRAMP: prediction of mammalian N6-methyladenosine (m6A) sites based on sequence-derived features. Nucleic Acids Res. 2016;44(10):e91.
- Liu B, Fang L, Wang S, et al. Identification of microRNA precursor with the degenerate K-tuple or Kmer strategy. J Theor Biol. 2015;385:153–159.
- Xiang S, Yan Z, Liu K, et al. AthMethPre: a web server for the prediction and query of mRNA m6A sites in Arabidopsis thaliana. Mol Biosyst. 2016;12(11):3333–3337.
- Li GQ, Liu Z, Shen HB, et al. TargetM6A: identifying N6-Methyladenosine sites from RNA sequences via position-specific nucleotide propensities and a support vector machine. IEEE Trans Nanobioscience. 2016;15(7):674–682.
- Huang Y, Niu B, Gao Y, et al. CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics. 2010;26(5):680–682.
- Vacic V, Iakoucheva LM, Radivojac P. Two sample logo: a graphical representation of the differences between two sets of sequence alignments. Bioinformatics. 2006;22(12):1536–1537.
- Bailey TL, Boden M, Buske FA, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–8.
- Wu X, Zhao W, Cui Q, et al. Computational screening of potential regulators for mRNA-protein expression level discrepancy. Biochem Biophys Res Commun. 2020;523(1):196–201.
- Wang X, Yan R, Li J, et al. SOHPRED: a new bioinformatics tool for the characterization and prediction of human S-sulfenylation sites. Mol Biosyst. 2016;12(9):2849–2858.
- Chen YZ, Tang YR, Sheng ZY, et al. Prediction of mucin-type O-glycosylation sites in mammalian proteins using the composition of k-spaced amino acid pairs. BMC Bioinformatics. 2008;9(1):101.
- Chen W, Zhang X, Brooker J, et al. PseKNC-General: a cross-platform package for generating various modes of pseudo nucleotide compositions. Bioinformatics. 2015;31(1):119–120.