
ABSTRACT
Despite the development of non-radioactive DNA/RNA labelling methods, radiolabelled nucleic acids are commonly used in studies focused on the determination of RNA fate. Nucleic acid fragments with radioactive nucleotide analoguesincorporated into the body or at the 5ʹ or 3ʹ terminus of the molecule can serve as probes in hybridization-based analyses of in vivo degradation and processing of transcripts. Radiolabelled oligoribonucleotides are utilized as substrates in biochemical assays of various RNA metabolic enzymes, such as exo- and endoribonucleases, nucleotidyltransferases or helicases. In some applications, the placement of the label is not a concern, while in other cases it is required that the radioactive mark is located at the 5ʹ- or 3ʹ-end of the molecule. An unsurpassed method for 5ʹ-end RNA labelling employs T4 polynucleotide kinase (PNK) and [γ-32P]ATP. In the case of 3ʹ-end labelling, several different possibilities exist. However, they require the use of costly radionucleotide analogues. Previously, we characterized an untypical nucleotidyltransferase named CutA, which preferentially incorporates cytidines at the 3ʹ-end of RNA substrates. Here, we demonstrate that this unusual feature can be used for the development of a novel, efficient, reproducible and economical method of RNA 3ʹ-end labelling by CutA-mediated cytidine tailing. The labelling efficiency is comparable to that achieved with the most common method applied to date, i.e. [5ʹ-32P]pCp ligation to the RNA 3ʹ-terminus catalysed by T4 RNA ligase I. We show the utility of RNA substrates labelled using our new method in exemplary biochemical assays assessing directionality of two well-known eukaryotic exoribonucleases, namely Dis3 and Xrn1.
Introduction
In recent years, many non-canonical terminal nucleotidyltransferases (NTases or TENTs) have been structurally, biochemically and functionally characterized [Citation1,Citation2]. Besides enzymes adding predominantly either adenosines or uridines, it becomes evident that various eukaryotic NTases display more intriguing substrate specificities [Citation3,Citation4].
Previously, we investigated molecular determinants underlying unique biochemical properties of fungal CutA NTase, which has a preference towards CTP, and explained them in the context of structural data, acquired through bioinformatics modelling and inspection of high-resolution crystal structures of the CutA catalytic core with RNA and NTP substrates [Citation5,Citation6]. In the course of our studies, we noted that CutA wild-type and mutant variants extend RNA oligonucleotide substrates by a limited number of nucleotides in the presence of non-labelled CTP or UTP. This prompted us to inquire whether CutA could be employed in a protocol for obtaining 3ʹ-labelled RNA substrates of defined length using [α-32P]CTP/UTP, as a reasonable alternative to existing methods.
RNA and DNA labelling techniques play an important role in molecular biology analytical methods and are often based on radioisotopes, such as 32P, 33P or 35S inserted into various sites of (deoxy)ribonucleotides. Labelled (d)NTPs can be incorporated into the body of nucleic acids or at their termini utilizing polymerases and DNA/RNA-modifying enzymes. In some applications (e.g. hybridization), the placement of the label is not particularly relevant. On the contrary, other kinds of experiments, such as in vitro characterization of biochemical activities of enzymes involved in RNA degradation or processing, require the use of oligonucleotide substrates labelled at either 5ʹ- or 3ʹ-end [Citation7–12]. Moreover, interpretation of data from biochemical assays is much more straightforward when labelled oligonucleotides of defined length are employed as substrates. Methods based on the use of e.g. template-independent polymerases, such as terminal deoxyribonucleotidyl transferase (TdT) or poly(A) polymerase (PAP) for 3ʹ-labelling of DNA or RNA molecules with [α-32P]dNTPs or NTPs, respectively, do not meet the latter criterion.
Radionucleotides are now increasingly substituted with fluorescently labelled counterparts, which are more stable over time and safer for the experimenter [Citation13]. However, large aromatic groups present in fluorophores may interfere with the enzymatic activity of some proteins, such as exoribonucleases. This often precludes the use of substrates with terminal fluorescent dyes in experiments aimed at the determination of the directionality of exonuclease action. The high cost of purchasing fluorescently labelled RNA molecules should also be taken into account, particularly when several substrates are intended to be tested in the course of planned experiments. Therefore, labelling techniques employing radioactive nucleotides are still widely utilized.
5ʹ-end RNA isotope labelling is usually achieved by using T4 PNK and [γ-32P]ATP and there is basically no other method that could outcompete this approach [Citation14,Citation15]. On the contrary, several methods for the generation of 3ʹ-radiolabelled RNA substrates of discrete length have been described. The most commonly used one is based on T4 RNA ligase 1 and [5ʹ-32P]pCp – a cytidine derivative radiolabelled at the 5ʹ terminus and with 3ʹ-terminal phosphate, which prevents extension of the reaction products beyond 1 nt [Citation16–18]. Alternatively, poly(A) polymerase can be used in conjunction with [α-32P]3′-deoxyadenosine (cordycepin) 5′-triphosphate (CoTP) – similar to pCp, the absence of free 3ʹ hydroxyl group on the ribose moiety of radiolabelled CoTP guarantees that no more than 1 nt is incorporated during labelling [Citation19,Citation20]. It should be emphasized that the efficiency of reactions catalysed by T4 RNA ligase 1 and poly(A) polymerase differs depending on the substrate length – the former works better for shorter RNA molecules, while the latter labels preferentially longer RNA species [Citation19]. Yet another option is the use of DNA polymerase I Klenow fragment and [α-32P]dATP after annealing of a short DNA template with 5ʹ-terminal NT- dinucleotide overhang to the RNA 3ʹ-end [Citation21].
Here, we show that the CutA NTase catalytic core purified to near homogeneity in a recombinant form can be successfully applied in a novel protocol of RNA 3ʹ-end labelling. The labelling efficiency is comparable to that achieved with the use of T4 RNA ligase I. Compared to the latter method, CutA also allows to generate labelled RNA molecules of defined length but extends substrates by two, rather than one, nucleotides. The combination of unmodified (wild-type) CutA and [α-32P]CTP as an NTP substrate is most advantageous regardless of the sequence present at the 3ʹ terminus of RNA oligonucleotides subjected to labelling. We further show that the labelling method based on CutA-mediated 3ʹ tailing proposed herein is applicable to a broad spectrum of substrates, including relatively short synthetic single-stranded oligoribonucleotides terminating at their 3ʹ-ends with different nucleotides, double-stranded oligonucleotides (both blunt-ended and equipped with 3ʹ single-stranded overhang on one of the strands) and several to over-a-dozen hundred nucleotides-long RNA molecules obtained by in vitro transcription. Finally, we demonstrate the utility of substrates labelled at the 3ʹ-end using this newly developed method in biochemical assays for the degradative activities of two eukaryotic exoribonucleases, triggering RNA decay from opposite termini – Dis3 and Xrn1.
Results and discussion
CutA adds two nucleotides to RNA substrate in the presence of either radiolabelled CTP or UTP
To test whether unique CutA enzymatic activity could be potentially employed for the development of a novel RNA 3ʹ-end labelling method, the catalytic core of the protein was purified to near homogeneity () as described in detail in Materials and Methods. A full representative course of CutA purification is presented in Fig. S1.
Figure 1. Activity of CutA WT and mutant variants on RNA oligonucleotide substrates of different length in the presence of radiolabelled CTP or UTP. (A) SDS-PAGE analysis of the CutA WT purified by two rounds of affinity chromatography on the Ni-NTA resin, followed by size exclusion chromatography. Sizes of the molecular weight marker (Perfect™ Colour Protein Ladder; EURx) bands are indicated on the right. (B) Labelling reactions. 20 nt-, 24 nt- or 31 nt-long oligoribonucleotides comprising identical 17 nt-long core sequence and A3, A7 or A14 3ʹ-terminal tail, respectively, were either labelled at the 5ʹ-end using T4 PNK and [γ-32P]ATP or incubated with equal amounts of CutA wild-type, N397A or R557H variant and [α-32P]CTP or [α-32P]UTP as a radioactive NTP substrate. RNA length is indicated on the left. 2 nt differences in length between 5ʹ-labelled substrates and reaction products generated by CutA variants are marked on the right
![Figure 1. Activity of CutA WT and mutant variants on RNA oligonucleotide substrates of different length in the presence of radiolabelled CTP or UTP. (A) SDS-PAGE analysis of the CutA WT purified by two rounds of affinity chromatography on the Ni-NTA resin, followed by size exclusion chromatography. Sizes of the molecular weight marker (Perfect™ Colour Protein Ladder; EURx) bands are indicated on the right. (B) Labelling reactions. 20 nt-, 24 nt- or 31 nt-long oligoribonucleotides comprising identical 17 nt-long core sequence and A3, A7 or A14 3ʹ-terminal tail, respectively, were either labelled at the 5ʹ-end using T4 PNK and [γ-32P]ATP or incubated with equal amounts of CutA wild-type, N397A or R557H variant and [α-32P]CTP or [α-32P]UTP as a radioactive NTP substrate. RNA length is indicated on the left. 2 nt differences in length between 5ʹ-labelled substrates and reaction products generated by CutA variants are marked on the right](/cms/asset/15a9ef78-4313-45d0-aa62-ef3269e002d9/krnb_a_1999104_f0001_b.gif)
Assays carried out previously during biochemical and structural characterization of CutA were done using RNA oligonucleotide substrates labelled at their 5ʹ termini and unlabelled NTPs. At high protein concentration, the enzyme was able to synthesize significantly extended tails, heterogeneous in length, particularly in the presence of ATP, but also CTP or UTP [Citation6]. This excluded the possibility of using CutA in the procedure of 3ʹ-end labelling, which would lead to the generation of substrates of defined size. However, decreasing protein concentration restricted the length of extensions added in the presence of CTP and UTP to 2 and 1 nt, respectively [Citation5,Citation6]. Furthermore, some mutant variants of CutA (e.g. N397A and R557H) appeared to add just one cytosine in the equivalent conditions [Citation5]. However, the efficiency of C- or U- tagging was largely dependent on the type of NTP used and nucleotide sequence present at the RNA substrate 3ʹ-end [Citation5].
We decided to validate whether CutA can be used for 3ʹ-end labelling of RNA oligonucleotides. Therefore we analysed the efficiency of labelling by several CutA variants and assessed the number of added nucleotides in in vitro assays with three substrates differing only in the length of 3ʹ-terminal A tails (3, 7 or 14 nt) using either [α-32P]CTP or [α-32P]UTP as NTP substrate. The same substrates were also labelled at their 5ʹ-ends with T4 PNK and [γ-32P]ATP, which produced 20 nt, 24 nt and 31 nt molecular weight markers. This allowed to estimate the length of an extension added by CutA in each of the tested conditions.
Reaction products were homogenous in length (). Labelling efficiency was significantly lower for R557H mutant compared to WT and N397A variants (). Most importantly, irrespective of the substrate, enzyme variant, and NTP used for labelling, RNA oligonucleotides were extended by 2 nucleotides at their 3ʹ termini (). Reproducible generation of reaction products of discrete length, extended by exactly two nucleotides, was encouraging in view of the potential use of CutA in the novel protocol of RNA 3ʹ-end labelling, different from the existing methods.
Combination of wild-type CutA and CTP is most favourable in terms of labelling substrates with different sequences at their 3ʹ ends
To further explore the possibility of using CutA as an alternative to T4 RNA ligase I, labelling was tested using two sets of RNA oligonucleotides. Within each set, the only difference was in the sequence present at the 3ʹ-end (22 nt RNA with A4 or U4 stretch and 17 nt RNA with A35 or A31U4 tail).
Since RNA substrates employed in the following series of experiments were equipped with FAM fluorophore at the 5ʹ-end, to ensure accurate interpretation of the results, it was necessary to verify beforehand whether such a large aromatic moiety affects migration of the substrate in the gel. Indeed, the products of reaction catalysed by wild-type CutA in the presence of 5ʹFAM-labelled 26 nt-long ss22-A4 oligoribonucleotide and [α-32P]CTP migrated in denaturing PAGE slower than for analogous substrate devoid of a fluorescent label at the 5ʹ-end – the difference was approximately 2 nucleotides (30 vs 28 nucleotides in the Fig. S2).
Experiments performed using just radiolabels, and substrates terminating with stretches of adenosines of different lengths or with 4 nt-long oligouridine tracts, allowed us to draw several important conclusions. First, ss22-U4 and ss17-A31U4 oligonucleotides were labelled significantly less efficiently than their counterparts containing only adenosines at their 3ʹ-ends, irrespective of NTP substrate and CutA variant (). Second, [α-32P]CTP proved to be a better donor of radioactivity than [α-32P]UTP (). Third, wild-type CutA outcompeted two remaining variants taking all tested combinations into account (). R557H mutant was barely productive, while N397A was significantly less efficient in labelling substrates terminating with uridines.
Figure 2. Efficiency of CutA-mediated RNA labelling and length of added extensions depends on the enzyme variant, 3ʹ-end RNA sequence and the radioactive NTP substrate. Two sets of RNA oligonucleotide substrates with FAM fluorophore at the 5ʹ-end: ss22-X4 (A,C) and ss17-A31X4 (B,D), terminating with stretches of adenosines or uridines were incubated with equal amounts of CutA wild-type, N397A or R557H variant and [α-32P]CTP/ [α-32P]UTP (marked as *CTP/*UTP above the lanes in A and B) or [α-32P]ATP (marked as *ATP in C and D) as a radioactive substrate. A: 20 nt-/24 nt-long and 22-/26-nt long size markers obtained by labelling of ss17-A3/ss17-A7 oligoribonucleotides at the 5ʹ-ends with T4 PNK and [γ-32P]ATP and at the 3ʹ-ends with CutA and [α-32P]CTP were run in parallel in leftmost and rightmost lanes, respectively. B,D: T4 PNK 5ʹ-radiolabelled ss17-A35 52-mer was run in utmost lanes as a size marker. C: T4 PNK 5ʹ-radiolabelled ss17-A14 31-mer was run in utmost lanes as a size marker. RNA length is indicated on both sides of the gels
![Figure 2. Efficiency of CutA-mediated RNA labelling and length of added extensions depends on the enzyme variant, 3ʹ-end RNA sequence and the radioactive NTP substrate. Two sets of RNA oligonucleotide substrates with FAM fluorophore at the 5ʹ-end: ss22-X4 (A,C) and ss17-A31X4 (B,D), terminating with stretches of adenosines or uridines were incubated with equal amounts of CutA wild-type, N397A or R557H variant and [α-32P]CTP/ [α-32P]UTP (marked as *CTP/*UTP above the lanes in A and B) or [α-32P]ATP (marked as *ATP in C and D) as a radioactive substrate. A: 20 nt-/24 nt-long and 22-/26-nt long size markers obtained by labelling of ss17-A3/ss17-A7 oligoribonucleotides at the 5ʹ-ends with T4 PNK and [γ-32P]ATP and at the 3ʹ-ends with CutA and [α-32P]CTP were run in parallel in leftmost and rightmost lanes, respectively. B,D: T4 PNK 5ʹ-radiolabelled ss17-A35 52-mer was run in utmost lanes as a size marker. C: T4 PNK 5ʹ-radiolabelled ss17-A14 31-mer was run in utmost lanes as a size marker. RNA length is indicated on both sides of the gels](/cms/asset/35b411ba-5dfc-4743-83a3-a572d08e17f8/krnb_a_1999104_f0002_b.gif)
Importantly, all radiolabelling reactions with [α-32P]CTP or [α-32P]UTP resulted in the extension of the substrates by exactly 2 nucleotides (). On the contrary, [α-32P]ATP led to the generation of the heterogeneous ladder of products (), which was inappropriate for RNA 3ʹ-end labelling.
Next, in an attempt to carefully and directly compare enzyme action in conditions when either limited amounts of radiolabels or saturating NTP concentration were used, as in our previous studies, parallel reactions were performed, using either exclusively trace amounts of [α-32P]CTP/UTP or a mixture of radionucleotide and an excess of its unlabelled counterpart (CTP or UTP, respectively).
Labelling reactions in the exclusive presence of radioactive CTP/UTP only led to substrate extension by 2 nucleotides in each tested case, as based on the radioactivity monitoring of labelled products. Notice that due to significant insufficiency of radioactive label compared to substrate concentration, no change in length at the level of fluorescence was detected, in stark contrast to reactions containing unlabelled NTP in addition to radionucleotides (Fig. S3). The excess of non-radioactive NTP usually led to the disappearance of the radiolabelled products (due to titration of the substrate; the only exception was 5ʹFAM-ss17-A31X4 substrates labelling with UTP; Fig. S3B). Analysis of fluorescent labels allowed to observe differences in the lengths of extensions depending on the RNA substrate, CutA variant and NTP utilized. In the presence of CTP, all four substrates were extended predominantly by 2 nucleotides by wild-type CutA (Fig. S3). N397A and R557H were able to add just one cytosine, but R557H was virtually inactive on substrates terminating with uridines (Fig. S3). In the presence of UTP, 5ʹFAM-ss22-A4 oligoribonucleotide was extended by only 1 nt. In turn, an addition of non-radioactive uridines to 5ʹFAM-ss22-U4 oligonucleotide was not detectable by fluorescence imaging for any of CutA variants (Fig. S3A). This was surprising, since trace amounts of radioactive UTP resulted in a clear, albeit sometimes not particularly efficient, addition of 2 nts to the 3ʹ-end of the fraction of these RNA substrates. Perhaps even more astonishingly, all CutA variants synthesized extensions comprising over a dozen nucleotides when UTP excess was used in the presence of ss17-A31X4 substrates. Interestingly, the radioactive label was incorporated with low efficiency into added oligouridine extensions (Fig. S3B). This suggests that longer tracts of adenosines present in the vicinity of RNA substrate 3ʹ-end substantially increase enzyme processivity towards uridines. Indeed, a more careful analysis performed using a set of ss22-An oligonucleotide derivatives, comprising n = 0, 1, 2, 4, 10 or 40 adenosines (Fig. S4) revealed that the threshold of oligo(A) length which stimulates enhanced enzyme processivity towards uridines is between 10 and 31 nt, considering all results presented herein.
The discrepancies between results of reactions carried out in the presence of traces of radionucleotides and excessive NTP concentration remain to be explained. Results of control reactions carried out with [α-32P]ATP as a ribonucleotide donor exclude the possibility that the radioactive label itself generally promotes reaction termination after an extension of the substrate by 2 nucleotides, since the wild-type enzyme is able to extend the substrate beyond this limit even when a trace of radioactive ATP is the only available NTP substrate for template-independent polymerization ( and Fig. S5).
The experiments described above clearly demonstrate that when radiolabelled CTP or UTP (unless the long A-tail is present at the substrate 3ʹ terminus), but not ATP, is the sole NTP substrate, wild-type CutA efficiently adds two nucleotides to every oligonucleotide tested, generating a product of a discrete length. This unexpected finding creates the basis for the potential use of these two agents in a novel 3ʹ-end RNA labelling method.
CutA-mediated labelling can be applied to a broad range of single-stranded RNA substrates with variable efficiency, dependent on the nature of the 3ʹ-terminal nucleotide
We wondered what is the spectrum of RNA substrates, which could be effectively labelled by CutA. All data presented so far were acquired mainly with the use of 5ʹFAM-ss22-A4 and 5ʹFAM-ss22-U4 substrates. We initially aimed to compare CutA-mediated radiolabelling results with previous detailed biochemical characterization of CutA enzymatic activity, which was achieved predominantly using the same oligoribonucleotides [Citation5,Citation6]. However, some aspects of 3ʹ-end labelling with radioactive NTPs (e.g. addition of two nucleotides instead of one in the presence of [α-32P]UTP) are quite unique compared to their non-radioactive counterparts. On the other hand, the general usability of the labelling method based on CutA activity proposed herein would be questionable if it was limited only to short single-stranded RNA (ssRNA) oligonucleotides with 3ʹ terminal adenosines or uridines. Therefore, we decided to more carefully examine the ability of CutA to label 3ʹ-ends of ssRNA oligonucleotides terminating with different nucleotides.
To this end, we extended the panel of tested substrates by other 5ʹFAM-labelled derivatives of the ss22 core oligonucleotide, which possessed an inherent single G (G1) at the 3ʹ-end: ss22-C4 and ss22-G4, terminating with stretches of C4 or G5, respectively, ss22-A1 (A1 at the 3ʹ-end) and ss22 (3ʹ-terminal G1) itself, as well as ss22-G1, ss22-C1 and ss22-C2 with 3ʹ-terminal G2, C1 and C2 nucleotides, respectively (schematic representation of all mentioned substrates is shown in the ; see Table S1 for nucleotide sequences).
Figure 3. CutA is able to label single-stranded RNA substrates with different 3ʹ termini, albeit with variable efficiency depending on the nucleotide composition of the ending. (A) Schematic representation of the utilized ssRNA substrates. Note the presence of terminal G inherently present in the ss22 core sequence common to all substrates within the tested set. Composition of the 3ʹ ending is provided on the right. (B) 5ʹFAM-labelled ss22-Xn substrates depicted in (A), with 3ʹ-ends containing different number of various nucleotides (as indicated at the bottom of each gel) were incubated with equal amounts of CutA WT and with a trace of radioactive [α-32P]CTP (*CTP; top) or [α-32P]UTP (*UTP; bottom), as indicated above the lanes. Both fluorescent and radioactive labels were detected following electrophoresis in the denaturing sequencing gel, as indicated on the right. 5ʹFAM-ss22, 5ʹFAM-ss22-A2 and 5ʹFAM-ss22-A4 as well as T4 PNK 5ʹ-radiolabelled ss17-A14 31-mer were run as fluorescent and radioactive size markers, respectively, in lanes next to the those with separated reaction products. Lengths of fluorescently labelled or radiolabelled oligonucleotides are indicated on the left
![Figure 3. CutA is able to label single-stranded RNA substrates with different 3ʹ termini, albeit with variable efficiency depending on the nucleotide composition of the ending. (A) Schematic representation of the utilized ssRNA substrates. Note the presence of terminal G inherently present in the ss22 core sequence common to all substrates within the tested set. Composition of the 3ʹ ending is provided on the right. (B) 5ʹFAM-labelled ss22-Xn substrates depicted in (A), with 3ʹ-ends containing different number of various nucleotides (as indicated at the bottom of each gel) were incubated with equal amounts of CutA WT and with a trace of radioactive [α-32P]CTP (*CTP; top) or [α-32P]UTP (*UTP; bottom), as indicated above the lanes. Both fluorescent and radioactive labels were detected following electrophoresis in the denaturing sequencing gel, as indicated on the right. 5ʹFAM-ss22, 5ʹFAM-ss22-A2 and 5ʹFAM-ss22-A4 as well as T4 PNK 5ʹ-radiolabelled ss17-A14 31-mer were run as fluorescent and radioactive size markers, respectively, in lanes next to the those with separated reaction products. Lengths of fluorescently labelled or radiolabelled oligonucleotides are indicated on the left](/cms/asset/21354cdf-781d-4ba0-b3b5-53c4db2e50c0/krnb_a_1999104_f0003_b.gif)
Interestingly, although substrates with terminal guanosines are known to be inefficiently modified by CutA in the presence of non-radioactive NTP due to some structural constraints that we had described earlier [Citation5], ss22-G4 was labelled to some extent by CutA in the presence of either radiolabelled CTP or UTP (). Similarly, 4 nt-long 3ʹ-terminal stretch of cytidines did not entirely preclude CutA-mediated labelling, although ss22-C4 appeared to be the least readily modified, particularly when [α-32P]UTP was used as a label donor (). Obviously, labelling efficiency for ss22-G4 and particularly ss22-C4 was significantly lower than for their counterparts ending with A4 or U4.
The labelling efficiency of ss22 terminating with a single G, particularly in the presence of radioactive CTP, was relatively high and comparable to ss22-A1 counterpart (). On the contrary, an extra G (ss22-G1) led to a significant drop of labelling efficacy. Similarly, C1 and C2 endings were labelled less efficiently than G1 in ss22 (). However, the effect was not as dramatic as in the case of C4. Weaker labelling of substrates containing pre-added cytidines (especially 4 in a row) is in agreement with the fact that CutA rarely synthesizes tails containing more than two consecutive cytidines [Citation6] – it probably usually detaches from the RNA molecule when the second cytidine is added.
Direct comparison of reaction products obtained in the presence of radiolabelled CTP or UTP disclosed better performance of the former as a radiolabel donor for all tested substrates, corroborating our previous observations (Fig. S6; compare to and to Fig. S3A,B).
Considering the number of cytidines added in the presence of radioactive CTP to RNA substrates with pre-existing terminal Cs, we would expect that the presence of a single C at the 3ʹ-end should theoretically promote the addition of only one extra C. However, this is apparently not the case, since in the presence of radioactive CTP traces such substrate is extended with 2 nucleotides, similarly to previously characterized ss22-A4 and ss22-U4 (). The exact calculation of the number of added nucleotides based on our new data with additional substrates was not straightforward since substrates with various 3ʹ endings displayed variabilities in gel migration, though containing the same number of nucleotides (e.g. 5ʹFAM-ss22-G1 migrated as 26 nt-long oligo, contrary to 5ʹFAM-ss22-A1, which migrated at the size of 25 nucleotides; also note differences in mobility between ss22-A4/ss22-U4/ss22-C4/ss22-G4 in the ). Nevertheless, direct comparison of patterns of electrophoretic migration of radioactively labelled products and fluorescently labelled substrates, which are virtually identical (), allows to conclude that the principle of ‘+2 nt extension’ is also true in the case of oligoribonucleotides terminating with C1, C2 and even C4.
Double-stranded substrates, both blunt-ended and with 3ʹ ssRNA overhang on one strand, are susceptible to 3ʹ-end labelling based on CutA-mediated tailing
To further expand the repertoire of RNA molecules, which can be 3ʹ-end-labelled by CutA, we employed double-stranded RNA (dsRNA) molecules by hybridizing different oligonucleotides belonging to 5ʹFAM-labelled ss22-Xn series with ss22compl RNA, i.e. oligoribonucleotide complementary to ss22 core sequence. By this means, we obtained either blunt- ended 5ʹFAM-ss22::ss22compl duplex or its derivatives with 3ʹ ssRNA overhangs on 5ʹFAM-labelled strand, representing various terminating nucleotides (). Completeness of the annealing was verified prior to labelling trials by comparing the migration of 5ʹFAM-labelled ssRNAs and corresponding duplexes in a native polyacrylamide gel. Visualized changes in electrophoretic mobility indicated that all fluorescently labelled strands were entirely incorporated into dsRNA substrates following annealing with ss22compl (Fig. S7).
Figure 4. Blunt and 3ʹ ss overhang-containing double-stranded RNA substrates can be 3ʹ-labelled by CutA-mediated tailing. (A) Schematic representation of the utilized dsRNA substrates, generated by hybridization of 5ʹFAM-ss22-Xn oligonucleotides with a complementary ss22compl strand. Composition of the 3ʹ ss overhang on the 5ʹFAM-labelled strand of the duplex is provided on the right. (B) 5ʹFAM-labelled ssRNA oligonucleotides and respective dsRNA substrates depicted in (A), with 3ʹ-ends/3ʹ ss overhangs containing different number of various nucleotides (as indicated below of each gel) were incubated with equal amounts of CutA WT and with a trace of radioactive [α-32P]CTP (*CTP; top) or [α-32P]UTP (*UTP; bottom), as indicated at the bottom of the gels. Both fluorescent and radioactive labels were detected following electrophoresis in the native polyacrylamide gel, as indicated on the right
![Figure 4. Blunt and 3ʹ ss overhang-containing double-stranded RNA substrates can be 3ʹ-labelled by CutA-mediated tailing. (A) Schematic representation of the utilized dsRNA substrates, generated by hybridization of 5ʹFAM-ss22-Xn oligonucleotides with a complementary ss22compl strand. Composition of the 3ʹ ss overhang on the 5ʹFAM-labelled strand of the duplex is provided on the right. (B) 5ʹFAM-labelled ssRNA oligonucleotides and respective dsRNA substrates depicted in (A), with 3ʹ-ends/3ʹ ss overhangs containing different number of various nucleotides (as indicated below of each gel) were incubated with equal amounts of CutA WT and with a trace of radioactive [α-32P]CTP (*CTP; top) or [α-32P]UTP (*UTP; bottom), as indicated at the bottom of the gels. Both fluorescent and radioactive labels were detected following electrophoresis in the native polyacrylamide gel, as indicated on the right](/cms/asset/384d34e6-f7bf-4ecf-96df-ae445864ed6a/krnb_a_1999104_f0004_b.gif)
To our surprise, even blunt-ended 5ʹFAM-ss22::ss22compl was prone to CutA-mediated 3ʹ tailing (). Labelling reaction performance was significantly better in the presence of radioactive CTP than UTP (Fig. S8). Such trend was observed for all tested substrates, both single- and double-stranded, but in the case of 5ʹFAM-ss22 superiority of [α-32P]CTP over [α-32P]UTP was exceptionally pronounced. 3ʹ ssRNA overhangs on 5ʹFAM-labelled strand composed of adenosines (A1, A2, A4, A10) significantly increased labelling efficiency both in the presence of radioactive CTP and UTP ( and Fig. S8). A similar but weaker effect was noted for U4 overhang, but only when [α-32P]CTP was used as an NTP substrate (). C4 overhang was the least efficiently labelled, in concordance with the results presented above for ssRNA substrates (; compare with and Fig. S6). The remaining overhangs (G4, G1, C1, C2) also decreased labelling efficacy in the presence of [α-32P]CTP (). Importantly, in most cases, variability in the labelling yield between dsRNAs containing different nucleotides within 3ʹ ss overhang recapitulate our previous findings made for corresponding ssRNA substrates with different 3ʹ endings ().
To verify which strand is labelled by CutA in the dsRNA substrates, we analysed the labelling products in the denaturing sequencing gel. Strikingly, only 5ʹFAM-labelled strand of the duplex was extended by CutA, whereas the complementary strand remained unmodified (Fig. S9). Otherwise, 24 nt-long labelled product would be observed in all lanes. The reason for such bias in labelling is unknown and we do not know whether this illustrates some commonly applicable rule. It could be envisaged that the large 5ʹFAM fluorophore present exactly opposite 3ʹ-hydroxyl group of the ss22compl oligonucleotide in the duplex somehow blocks its labelling. Potential users-to-be of the enzyme should bear in mind that labelling of the selected duplex strand can probably be enforced in other cases by chemical blocking of the complementary strand 3ʹ-end, e.g. with phosphate or amino group.
The proven ability of CutA to label double-stranded RNA molecules opens up the possibility to use the RNAs labelled in this way in biochemical studies of enzymes, which are active towards more complex substrates, e.g. RNA helicases.
Not only short synthetic oligoribonucleotides, but also much longer in vitro transcribed mRNAs can be 3ʹ-labelled with CutA
Comprehensive biochemical characterization of RNA metabolic enzymes often requires demonstration of their catalytic activities on substrates more physiologically relevant than synthetic oligoribonucleotides, i.e. specific transcripts present in the cellular environment. Therefore, we performed additional labelling experiments using in vitro transcribed mRNAs instead of short RNA oligonucleotides.
Two mRNAs differing in length, encoding Gaussia princeps luciferase (GLuc; 869 nt) and Photinus pyralis luciferase (FLuc; 1974 nt) were synthesized by in vitro transcription, each in two variants – lacking (pA−) or containing (pA+) 36 nt-long poly(A) tail (i.e. lengths of GLuc pA+ and FLuc pA+ were 905 nt and 2010 nt, respectively), as described in Materials and Methods. These transcripts were then labelled with CutA using either [α-32P]CTP or [α-32P]UTP as an NTP substrate and separated in both native and denaturing agarose gels. Ethidium bromide staining of the native gel revealed migration of all synthesized and labelled transcripts at approximately correct sizes when compared to RNA ladder run in parallel (; top). Separated mRNAs were transferred from both gels onto nylon membranes to avoid diffusion and enable visualization of the radioactively labelled transcripts. Staining of the membranes followed by phosphorimaging confirmed that all four transcripts were labelled at the 3ʹ-end with CutA (). The identity of the transcripts after transfer was additionally examined by northern blot hybridization with complementary DNA probes (; bottom). Altogether, similar to what we found previously in the case of synthetic oligoribonucleotide substrates, labelling efficiency for much longer, in vitro transcribed mRNAs, is also largely dependent on the 3ʹ-terminal sequence present in the substrate and the radiolabelled NTP substrate used. First, GLuc and FLuc pA+ substrates are labelled more efficiently than their pA− counterparts terminating with 3 uridines due to cleavage of the plasmid template for IVT with PmeI restriction endonuclease, which recognizes GTTT^AAAC palindrome. Second, [α-32P]CTP performs better than [α-32P]UTP as a radiolabel donor, particularly in the case of pA+ transcripts ().
Figure 5. Longer RNA molecules, such as mRNAs obtained by the in vitro transcription are prone to 3ʹ-end labelling based on CutA enzymatic activity. Non-polyadenylated (pA−) or polyadenylated (pA+) versions of transcripts coding for G. princeps (GLuc) and P. pyralis (FLuc) luciferases were synthesized by in vitro transcription on plasmid templates linearized with appropriate restriction endonucleases. Following purification, mRNAs were incubated with equal amounts of CutA WT and with a trace of radioactive [α-32P]CTP (*CTP) or [α-32P]UTP (*UTP), as indicated at the bottom of the gels, and separated in either native, ethidium bromide-stained (top) or in denaturing (bottom) agarose gel. Transcripts were then immobilized on nylon membranes, which were stained with methylene blue. After staining, radioactive signal from labelled mRNAs was detected by phosphorimaging. Membrane corresponding to denaturing gel was additionally hybridized with northern blot probes specific to FLuc and GLuc mRNAs. Solid arrowheads in northern blot panels designate signals, which are aggregates of CutA-mediated labelling and indicated probe hybridization. Residual radioactive signals from transcripts labelled with CutA are marked with open arrowheads. Sizes of the molecular weight marker (RiboRulerTM High Range RNA Ladder), which was run in the leftmost lane, are marked on the left
![Figure 5. Longer RNA molecules, such as mRNAs obtained by the in vitro transcription are prone to 3ʹ-end labelling based on CutA enzymatic activity. Non-polyadenylated (pA−) or polyadenylated (pA+) versions of transcripts coding for G. princeps (GLuc) and P. pyralis (FLuc) luciferases were synthesized by in vitro transcription on plasmid templates linearized with appropriate restriction endonucleases. Following purification, mRNAs were incubated with equal amounts of CutA WT and with a trace of radioactive [α-32P]CTP (*CTP) or [α-32P]UTP (*UTP), as indicated at the bottom of the gels, and separated in either native, ethidium bromide-stained (top) or in denaturing (bottom) agarose gel. Transcripts were then immobilized on nylon membranes, which were stained with methylene blue. After staining, radioactive signal from labelled mRNAs was detected by phosphorimaging. Membrane corresponding to denaturing gel was additionally hybridized with northern blot probes specific to FLuc and GLuc mRNAs. Solid arrowheads in northern blot panels designate signals, which are aggregates of CutA-mediated labelling and indicated probe hybridization. Residual radioactive signals from transcripts labelled with CutA are marked with open arrowheads. Sizes of the molecular weight marker (RiboRulerTM High Range RNA Ladder), which was run in the leftmost lane, are marked on the left](/cms/asset/8d5ed525-3456-4a26-8233-4d5bdb2b5d1b/krnb_a_1999104_f0005_b.gif)
The ability of CutA to label 3ʹ-ends of several to over-a-dozen hundred-long substrates, particularly mRNAs, may be useful e.g. in biochemical in vitro studies focusing on transcript properties depending on the 5ʹ-end modifications (phosphorylation status, different types of 5ʹ cap structures etc.).
Efficiency of oligoribonucleotide labelling with CutA and [α-32P]CTP is similar to 3ʹ-end [5ʹ-32P]pCp ligation catalysed by T4 RNA ligase I
To directly assess the efficiency of the labelling reaction with CutA and radiolabelled CTP or UTP in relation to the most commonly 3ʹ-end labelling method, i.e. T4 RNA ligase I-mediated ligation of [5ʹ-32P]pCp, we compared these two reactions using the same amounts of identical RNA substrates, similar concentrations of proteins and their respective radionucleotide substrates.
First, amounts of CutA WT were carefully adjusted to levels comparable to commercial T4 RNA ligase I (New England Biolabs) (). Besides, two CutA preparations – fresh one or stored for 1 year at 4°C – were analysed alongside to assess protein stability and their activities directly collated ( and Fig. S10). SDS-PAGE analysis disclosed the presence of additional bands in the CutA WT sample after prolonged storage as compared to a freshly purified enzyme, most likely representing degradation products. Despite slight proteolysis, both preparations had similar NTase activity when assayed with ATP, CTP or UTP (Fig. S10), indicating that the enzyme is relatively stable and retains its properties even after long-term storage in suboptimal conditions.
Figure 6. Comparison of RNA 3ʹ-end labelling methods employing CutA and T4 RNA ligase 1. (A) SDS-PAGE analysis of adjusted amounts of two CutA preparations (freshly purified and stored for 1 year at 4°C) and T4 RNA ligase 1 from NEB. (B) and (C) Schemes of reactions catalysed by CutA in the presence of [α-32P]CTP (A) and by T4 RNA ligase 1 in the presence of [5ʹ-32P]pCp (B), leading to k + 2 nt-long (A) and k + 1 nt-long (B) products from k nt-long substrate, respectively. Upper part of panel B (grey colour) shows the optional possibility of [5ʹ-32P]pCp self-preparation by T4 PNK-mediated 5ʹ-end labelling of 3ʹ-CMP. (D) Large-scale labelling of 5ʹ-phosphorylated and 5ʹ-hydroxyl ss17-A14 RNA oligonucleotides with T4 RNA ligase 1 and [5ʹ-32P]pCp or with CutA WT and [α-32P]CTP/UTP. Equal amounts of proteins and radionucleotides were used. Reaction products were separated in preparative denaturing PAGE and exposed to an X-ray film which was developed prior to excision of gel fragments containing labelled oligoribonucleotides. (E) Analysis of gel-purified reaction products from (D), together with T4 PNK 5ʹ-labelled ss17-A14 RNA oligonucleotide (leftmost lane) in low- (top) and high-resolution (bottom) gels. Approximately equal amounts of radioactivity (based on handheld radioactivity detector counting) were loaded into each lane
![Figure 6. Comparison of RNA 3ʹ-end labelling methods employing CutA and T4 RNA ligase 1. (A) SDS-PAGE analysis of adjusted amounts of two CutA preparations (freshly purified and stored for 1 year at 4°C) and T4 RNA ligase 1 from NEB. (B) and (C) Schemes of reactions catalysed by CutA in the presence of [α-32P]CTP (A) and by T4 RNA ligase 1 in the presence of [5ʹ-32P]pCp (B), leading to k + 2 nt-long (A) and k + 1 nt-long (B) products from k nt-long substrate, respectively. Upper part of panel B (grey colour) shows the optional possibility of [5ʹ-32P]pCp self-preparation by T4 PNK-mediated 5ʹ-end labelling of 3ʹ-CMP. (D) Large-scale labelling of 5ʹ-phosphorylated and 5ʹ-hydroxyl ss17-A14 RNA oligonucleotides with T4 RNA ligase 1 and [5ʹ-32P]pCp or with CutA WT and [α-32P]CTP/UTP. Equal amounts of proteins and radionucleotides were used. Reaction products were separated in preparative denaturing PAGE and exposed to an X-ray film which was developed prior to excision of gel fragments containing labelled oligoribonucleotides. (E) Analysis of gel-purified reaction products from (D), together with T4 PNK 5ʹ-labelled ss17-A14 RNA oligonucleotide (leftmost lane) in low- (top) and high-resolution (bottom) gels. Approximately equal amounts of radioactivity (based on handheld radioactivity detector counting) were loaded into each lane](/cms/asset/50eb74f6-206f-43d1-824d-72a6c20fde7a/krnb_a_1999104_f0006_b.gif)
The schemes of radiolabelling reactions catalysed by CutA and T4 RNA ligase I are juxtaposed in , respectively. As documented above, CutA adds 2 nucleotides to k nt-long oligonucleotide substrate in the presence of radioactive pyrimidine triphosphate (e.g. CTP), thus leading to k+2 nt-long product (). In the case of T4 RNA ligase, the most commonly used substrate is [5ʹ-32P]pCp – a modified cytidine containing radiolabelled phosphate at the 5ʹ-end and another phosphate at the 3ʹ-end, blocking ligation after a single round of reaction. [5ʹ-32P]pCp is commercially available in a ready-to-use form but can also be self-prepared by labelling of 3ʹ cytidine monophosphate (3ʹ-CMP) with T4 polynucleotide kinase and [γ-32P]ATP (, upper part). Ligation of [5ʹ-32P]pCp to the hydroxyl group at the 3ʹ-end of k nt-long substrate generates k+1 nt-long product ()).
To compare efficiency of the two labelling methods, reactions were carried out using identical amounts (50 µCi) of respective radionucleotides ([5ʹ-32P]pCp for T4 RNA ligase I and [α-32P]CTP or UTP for CutA WT) and 1 µM ss17-A14 RNA oligonucleotide in the presence of molar equivalents of appropriate enzymes. Since we planned to assess the utility of labelled substrates in degradation assays employing i.a. Xrn1 exoribonuclease, which is sensitive to 5ʹ-end phosphorylation status, two variants of the substrate were used: either containing (5ʹP-ss17-A14) or lacking (5ʹOH-ss17-A14) 5ʹ-phosphate. Entire volumes of all reactions containing reaction products were resolved in preparative denaturing PAGE. Following electrophoresis, the gel was exposed to a film, which demonstrated that the signal corresponding to products of reactions catalysed by T4 RNA ligase I and CutA WT in the presence of radiolabelled CTP were of similar intensity (). When radioactive UTP was used instead as CTP as a substrate for CutA WT, the signal was less intense, in agreement with observations made for other substrates, which indicated that UTP is a less optimal substrate for labelling (; see also , , Fig. S3, Fig. S6 and Fig. S8). Products of all labelling reactions were subsequently gel-purified. Analysis of labelled oligoribonucleotides demonstrated the expected differences in length (), resulting from distinct reaction mechanisms for T4 RNA ligase I and CutA WT, described above and depicted in , respectively.
In summary, we found that the labelling efficiency achieved for CutA WT in the presence of [α-32P]CTP is comparable to that obtained for T4 RNA ligase I and its most commonly applied substrate, [5ʹ-32P]pCp. This opened the possibility of testing the usefulness of oligoribonucleotide substrates labelled using CutA WT in the exoribonuclease degradation assays.
Degradation assays with exoribonucleases working in the opposite directions demonstrate the utility of substrates labelled at the 3ʹ-end with the use of CutA
To demonstrate the usefulness of RNA oligonucleotide substrates labelled at the 3ʹ-end employing CutA in downstream biochemical analyses, degradation assays with the use of two distinct yeast exoribonucleases, Dis3 3ʹ-5ʹ exonuclease and Xrn1 5ʹ-3ʹ exonuclease, were performed. For this purpose, degradation of ss17-A14 oligoribonucleotides, bearing 5ʹ-hydroxyl or 5ʹ-phosphate, labelled at the 3ʹ-end by T4 RNA ligase I-mediated [5ʹ-32P]pCp ligation, or their counterparts prepared using CutA WT and [α-32P]CTP/UTP, was assessed in parallel. Additionally, ss17-A14 RNA oligo labelled at the 5ʹ terminus with T4 PNK and [γ-32P]ATP was employed as a control.
In line with what is known about Dis3 exonuclease activity [Citation22,Citation23], the enzyme degraded 5ʹ-labelled substrate to 3–4 nt-long oligonucleotide products, independently of the 5ʹ phosphorylation status (, left part; Fig. S11A). 3ʹ-labelled substrates, irrespective of which enzyme was used for substrate preparation, were degraded to mononucleotides. Notably, a signal from 1 nt-long products was more intense for substrates labelled using CutA than T4 RNA ligase I (, left part; Fig. S11A). Analysis of the reaction products in lower resolution gel revealed that this is probably due to the fact that released p*Cp is likely decomposed to free radioactive phosphate, which migrates faster than the single nucleotide (Fig. S11B).
Figure 7. The use of RNA oligonucleotide substrates labelled with CutA at the 3ʹ-end in exoribonuclease degradation assays. Activity assays for Dis3 3ʹ-5ʹ and Xrn1 5ʹ-3ʹ exoribonucleases were performed using ss17-A14 RNA oligonucleotide substrates labelled at either 5ʹ- or 3ʹ-end employing various methods. Note that in the case of Xrn1, 5ʹ-phosphorylated substrates labelled at the 3ʹ-end were used due to the fact that Xrn1 is sensitive to phosphorylation status of the 5ʹ RNA terminus. Reactions were terminated after the time points indicated above the lanes and the products were separated by high-resolution PAGE. RNA length is indicated on the left
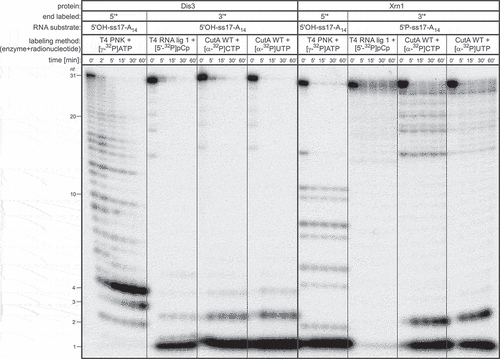
In the case of Xrn1, degradation of 5ʹ-labelled substrate produced mononucleotides (, right part; Fig. S12A). For 3ʹ-labelled substrates, the ability of Xrn1 to promote their decay was strictly dependent on the presence of 5ʹ-terminal phosphate (Fig. S12A). Incubation of 5ʹ-phosphorylated RNA oligonucleotides labelled at the 3ʹ-end with CutA and [α-32P]CTP/UTP resulted in the generation of predominantly mononucleotides and a minor fraction of dinucleotides (, right part; Fig. S12A). On the contrary, 1 nt-long products for the analogous substrate with 3ʹ-label added by T4 RNA ligase I were hardly visible in high-resolution gel (Fig. S12A). Electrophoresis in low-resolution gel was mandatory to monitor the fate of these mononucleotide products, which were apparently degraded further to free phosphate and this effect was even more pronounced than in the case of Dis3-mediated 3ʹ-5ʹ exoribonucleolytic degradation (Fig. S12B; compare with Fig. S11B).
To summarize, RNA oligonucleotide 3ʹ labelling by CutA and [α-32P]CTP/UTP is a superior alternative to T4 RNA ligase 1-mediated [5ʹ-32P]pCp ligation, and labelled substrates can be employed in biochemical assays aiming at analysis of enzymatic properties of various exoribonucleases, such as assessment of their directionality. Moreover, interpretation of data obtained using CutA-labelled oligoribonucleotides proves to be easier, since the results are more clear due to the fact that final mononucleotide products are not further disintegrated to phosphates, as is observed for corresponding substrates with 3ʹ-label added by T4 RNA ligase 1.
Conclusion
Following the simple protocol described in this report, homogenous and monomeric CutA protein fragment encompassing catalytic core can be obtained in high amounts after two-step nickel affinity chromatography and gel filtration. We show that CutA thus obtained is enzymatically active and retains its properties even after prolonged storage at suboptimal temperature. The procedure described herein should be easily adaptable to available resources and feasible in every laboratory having a basic knowledge of protein purification.
We are convinced that our CutA-based protocol can be considered as nearly universal for in vitro 3ʹ-end labelling of any RNA molecule. We have demonstrated that all substrates tested herein, from short single-stranded RNAs with different 3ʹ-endings (in terms of nucleotide composition and length) and RNA duplexes (either blunt or with 3ʹ ss overhangs) to considerably longer mRNAs, are susceptible to CutA-mediated tailing. Obviously, labelling efficiencies vary, being dependent mainly on the nature of the 3ʹ-terminal nucleotides and on the radioactive NTP used as a radiolabel donor. Nonetheless, even though seemingly lower labelling efficacy was shown in our report for some RNAs, like those terminating with cytosines and guanosines, it does not disqualify CutA-mediated tailing as a method of choice for such substrates. From the technical point of view, reduced efficiency of radionucleotide incorporation can be easily compensated for by increasing the label concentration in the reaction mixture or by decreasing the final volume in which the final labelling product is recovered (e.g. the volume in which the pellet of labelled oligonucleotide obtained after isopropanol precipitation from the gel eluate after preparative PAGE is resuspended).
In several aspects, the method of 3ʹ-end labelling presented herein is superior to the approaches used so far. First, our experiments with exoribonuclease-mediated degradation of 3ʹ-labelled RNA substrates show that the mononucleotide decay products are better visible for oligonucleotides labelled with CutA WT and [α-32P]CTP than with T4 RNA ligase 1 and [5ʹ-32P]pCp, particularly for Xrn1, initiating RNA degradation from the 5ʹ terminus. Since no decomposition of 1 nt-long product to radioactive phosphate occurs, the results of biochemical assays are cleaner and their interpretation is more straightforward. Second, T4 RNA ligase 1-mediated ligation of pCp is efficient provided that high concentrations of both enzyme and radioisotope are utilized. Moreover, manufacturers and protocol creators suggest that overnight incubation at lower temperatures prevents RNA substrate degradation during ligation. In contrast, the labelling reaction catalysed by CutA WT is complete within 1–2 hours of incubation at 37°C. In addition, [α-32P]CTP/UTP is currently considerably cheaper and more readily available radioactivity donor than [5ʹ-32P]pCp or – particularly – [α-32P]CoTP, required in alternative labelling methods, depending on the use of T4 RNA ligase I or poly(A) polymerase. Finally, some exonucleases, like for instance RRP6, are inhibited by 3ʹ terminal phosphate, which is present in substrates labelled using pCp [Citation24].
We believe that in addition to biochemical analyses of RNA metabolic enzymes, substrates labelled at the 3ʹ-end with CutA can be used in other applications, e.g. RNA structure probing [Citation25], footprinting [Citation26] or EMSA [Citation27,Citation28].
Materials and Methods
CutA expression, purification and storage conditions
E. coli BL21-CodonPlus(DE3)-RIL strain (Agilent; E. coli B F – ompT hsdS[rB – mB–] dcm+ Tetr gal λ[DE3] endA Hte [argU ileY leuW Camr]) was transformed with pCC25 plasmid containing sequence encoding wild-type Thielavia terrestris CutA protein fragment corresponding to amino acids 240–603, generated by sequence- and ligation-independent cloning (SLIC) into BamHI-XhoI sites of pET28M N-6xHis-SUMOTag vector, or with pCC33/pCC37 plasmids for expression of N397A/R557H variants of the protein [Citation5]. Transformants were then grown in standard Luria-Broth (LB) medium with 50 µg/ml kanamycin and 34 µg/ml chloramphenicol. This starter culture was subsequently used to inoculate 1 l of Auto Induction Medium (AIM) Super Broth Base including Trace elements (Formedium) with 2% glycerol and both antibiotics. Bacteria were then grown for 48 h at 18°C with shaking (150 rpm) and collected by centrifugation at 4500 rpm in a Sorvall H6000A/HBB6 swinging-bucket rotor for 15 min at 4°C.
Bacterial pellet was re-suspended in 70 ml of lysis buffer (50 mM Tris-HCl pH 7.4, 50 mM NaCl, 10 mM imidazole, 10 mM 2-mercaptoethanol, 1 mM phenylmethylsulfonyl fluoride [PMSF]), incubated with lysozyme (50 µg/ml; Roth) for 30 min in a cold room, and then broken in an EmulsiFlex-C3 High Pressure homogenizer at 1500 Bar. The homogenate was centrifuged in a Sorvall WX Ultra Series ultracentrifuge (F37L rotor) at 32,000 rpm for 45 min at 4°C.
A representative course of CutA purification is presented in the Fig. S1. Fractions from different stages of purification procedure were analysed by standard SDS-PAGE. N-terminally His-SUMO-tagged enzyme was efficiently produced in bacteria following auto induction and displayed high solubility (see lane EX in the Fig. S1C).
The extract was used for protein purification using the ÄKTA Xpress system (GE Healthcare), employing nickel affinity chromatography on an ÄKTA-compatible 5 ml column that was filled with Ni-NTA Superflow resin (Qiagen). The column was equilibrated with 25 ml of LS buffer before extract loading. After protein binding, the resin was sequentially washed with 40 ml of low-salt (LS) buffer (50 mM Tris-HCl pH 7.4, 50 mM NaCl, 10 mM imidazole), 25 ml of high-salt (HS) buffer (50 mM Tris-HCl pH 7.4, 1 M NaCl, 10 mM imidazole), and 20 ml of LS buffer. Bound proteins were recovered by elution with 30 ml of buffer E (50 mM Tris-HCl pH 7.4, 50 mM NaCl, 300 mM imidazole). This first round of purification on the nickel column usually resulted in two peaks after elution, most probably due to column overload (Fig. S1A). Pooled fractions representing the first peak (black bar EL1 in the Fig. S1A and corresponding lane in the Fig. S1C) were dialysed against 2 l of LS buffer in the presence of 50 µg of home-made SUMO protease. Analysis of the reaction mixture after dialysis indicated that the cleavage was almost complete, leading to the release of untagged CutA (lane DIAL in the Fig. S1C).
This mixture was then subjected to second round of purification on the nickel resin, performed using ÄKTA Purifier system (GE Healthcare) and employing LS buffer for collection of the flow-through and buffer E2 (50 mM Tris-HCl pH 7.4, 1 M NaCl, 300 mM imidazole) for elution. This resulted in separation of CutA (black bar FT2 in the Fig. S1B and corresponding lane in the Fig. S1C) in the flow-through from His-tagged SUMO protease and cleaved-off His-SUMO tag (black bar EL2 in the Fig. S1B and corresponding lane in the Fig. S1C), which re-bound to the column and were eluted after increase in the imidazole concentration. Notably, fraction FT2 after two rounds of affinity chromatography contained desired full-length protein of relatively high purity, contaminated only with bacterial chaperones and degradation products.
Further purification of 1 ml of pooled FT2 fractions (less than 1/10th of available material) by gel filtration on size exclusion Superdex 75 10/300 GL column (GE Healthcare) using 1.2 column volumes of gel-filtration (GF) buffer (20 mM Tris-HCl pH 7.4, 50 mM NaCl) resulted in asymmetric peak (marked with black bar in the Fig. S1D). Analysis of fractions covered by this peak (Fig. S1E) showed that its left part corresponded to CutA contaminated with some chaperones (and maybe partially aggregated), while the prevailing right part contained largely intact protein – minor degradation products (ca. 25 and 15 kDa) were present already in the input prior to size exclusion chromatography (lane FT2 in the Fig. S1C) and apparently difficult to be separated from the full-length CutA. Protein concentration in the final CutA preparation obtained from 1 litre of bacterial culture after all purified steps described above (pooled fractions A9-A10 after gel filtration – ca. 800 µl) was approximately 2 mg/ml. Molecular weight of the purified CutA employed in biochemical analyses and labelling experiments presented herein was 41.84 kDa (close to the theoretical mass of the protein, i.e. 42.23 kDa), as calculated by SEC-MALS analysis (Fig. S1F; see below for technical details).
Although CutA appears to be stable and retains activity even after prolonged storage at 4°C, we recommend addition of glycerol to final concentration of 30%, division into aliquots, flash-freezing in liquid nitrogen and storage at −80°C.
SEC-MALS
Size exclusion chromatography coupled to multi-angle light scattering (SEC-MALS) analysis was performed using a high-performance liquid chromatography (HPLC) instrument (1260 Infinity LC, Agilent) equipped with a UV detector, MALS detector (DAWN HELEOS II, Wyatt Technology) and a differential refractometer (Optilab T-rEX, Wyatt Technology). 100 µl of purified CutA (2 mg/ml) was loaded onto a Superdex 200 Increase 10/300 column (GE Healthcare) equilibrated with GF buffer. The sample was run at room temperature at a flow rate of 0.5 ml/min. UV wavelengths of 280 nm and 254 nm were used for sample monitoring during SEC. The results were analysed using ASTRA v.6 software (Wyatt Technology) in accordance with manufacturer’s instructions.
Nucleotidyltransferase assays
The PAGE-purified RNA oligonucleotides with fluorescein amidite (FAM) modification at the 5′-end (see Table S1 for nucleotide sequences) were purchased from Future Synthesis (Poznan, Poland; http://futuresynthesis.com). In vitro nucleotidyltransferase reactions were performed in a 20 µl volume using buffer R that contained 10 mM Tris-HCl pH 8.0, 50 mM NaCl, 1 mM MnCl2, 1 mM DTT, and either no NTP or 1 mM ATP, CTP or UTP. 1 µM of CutA and 1 µM of 5′ FAM-labelled oligoribonucleotide substrate was used. The reactions were performed at 37°C and stopped at various time-points by collecting an aliquot of the reaction mixture and adding it to an equal volume of formamide gel loading dye (90% formamide, 20 mM EDTA, 0.03% bromophenol blue, and 0.03% xylene cyanol in 1x Tris-borate-EDTA [TBE]), followed by flash freezing in liquid nitrogen. Following thermal denaturation at 90°C for 5 min, reaction products were resolved either in denaturing 20% low-resolution polyacrylamide/8 M urea/1x TBE gels or in 10% sequencing PAGE if single-nucleotide resolution was required. A Typhoon FLA 9000 laser scanner (GE Healthcare) was used to visualize fluorescently labelled reaction products.
Preparation of double-stranded RNA (dsRNA) molecules and validation of annealing
Hybridization of individual 5ʹFAM-ss22-Xn oligonucleotides with complementary ss22compl strand (see Table S1 for nucleotide sequence) was achieved by mixing 80 µl of the annealing buffer (10 mM Tris-HCl pH 7.5, 50 mM NaCl) with 10 µl of each of the two oligoribonucleotides (100 µM), incubating the mixture for 7 minutes at 99°C in a thermoblock and switching off the device until the samples cooled down to the room temperature. This resulted in the generation of 10 µM duplex stock solutions. Proper annealing of dsRNA strands was verified by parallel analysis of ssRNAs and generated duplexes in 15% native polyacrylamide/1xTBE gel. Glycerol loading dye (50% glycerol, 20 mM EDTA, 0.025% bromophenol blue, and 0.025% xylene cyanol in 1xTBE) was used for sample preparation before electrophoresis in native conditions.
Generation of mRNA substrates by in vitro transcription (IVT)
GLuc and FLuc mRNAs were obtained by in vitro transcription reaction using pJET-based constructs, encoding either G. princeps luciferase [Citation29] or P. pyralis luciferase, as templates (both kindly gifted by Dr Pawel Sikorski from the Centre of New Technologies, Warsaw, Poland). In both cases, non-polyadenylated (pA−; lacking polyA tail) and polyadenylated (pA+; containing 36 nt-long polyA tail) variants were synthesized using plasmids linearized with PmeI or AarI restriction enzyme (both from Thermo Fisher Scientific), respectively. IVT reactions were set in 20 µl volume containing RNA polymerase buffer (40 mM Tris-HCl pH 7.9, 25 mM MgCl2, 1 mM DTT, 2 mM spermidine), 40 ng/μl of linearized DNA template; NTPs (5 mM each; Thermo Fisher Scientific), 0.002 U/µl pyrophosphatase, inorganic (Thermo Fisher Scientific) and 2 µl of home-made T7 RNA polymerase. Mixtures were incubated at 37°C for 1 h. Afterwards, additional 2 µl of T7 RNA polymerase were added and reactions were continued for another 1 h at 37°C. Next, DNase I (Thermo Fisher Scientific) was added (0.1 U/µl) and samples were incubated at 37°C for 30 minutes. Crude mRNAs were purified using Monarch RNA Cleanup Kit (New England Biolabs).
Labelling of RNA substrates with radioactive nucleotides
5ʹ-end labelling of oligoribonucleotides (see Table S1 for nucleotide sequences) was performed at 37°C for 90 minutes using 10 units of T4 polynucleotide kinase (PNK; NEB) and 3 µCi of [γ-32P]ATP (Hartmann Analytic; SRP-401) in 10 µl reaction volume containing 1 µM RNA substrate.
3ʹ-end small-scale test labelling of single-stranded RNA (ssRNA) oligonucleotides or respective dsRNA substrates with CutA was performed at 37°C for 90 minutes in 20 µl reaction volume containing buffer R, 1 µM enzyme, 1 µM RNA substrate, 20 units of RiboLock RNase Inhibitor (Thermo Fisher Scientific) and 3 µCi (1 pmol) of [α-32P]CTP (Hartmann Analytic; SRP-209), [α-32P]UTP (Hartmann Analytic; SRP-210) or [α-32P]ATP (Hartmann Analytic; SRP-207). Where indicated, respective unlabelled NTPs were added to reaction mixtures at 1 mM concentration. Following radiolabelling reactions, the products were separated in either denaturing 20% low-resolution/10% sequencing gels or in native 15% gels, as described above. Fluorescent and radioactive labels were visualized using whichever of Typhoon FLA 9000/9500/7000 scanners available.
Small-scale labelling of in vitro transcribed GLuc and FLuc mRNAs with CutA was performed as in the case of synthetic oligoribonucleotides, but using 7.5 pmol of the RNA substrate per 20 µl of reaction. Single extraction with acidic phenol (pH 4.3):chloroform (1:1, v:v) was performed to get rid of protein and to facilitate preservation of mRNA integrity.
Large scale 3ʹ-end labelling of ssRNA oligonucleotide substrates with CutA was performed at 37°C for 90 minutes in 20 µl reaction volume containing buffer R, 2.5 µM enzyme, 1 µM RNA substrate, 20 units of RiboLock RNase Inhibitor and 50 µCi of [α-32P]CTP or [α-32P]UTP.
Large scale 3ʹ-end labelling of ssRNA oligonucleotide substrates with T4 RNA ligase 1 was performed at 16°C overnight using 20 units of T4 RNA ligase 1 (ssRNA ligase; NEB) and 50 µCi of [5ʹ-32P]pCp in 20 µl reaction volume containing 1x reaction buffer (NEB), 1 µM oligoribonucleotide, 1 mM ATP, 10 mM DTT, 10% (v/v) DMSO and 20 units of RiboLock RNase Inhibitor.
Large scale labelling reaction products were subjected to phenol:chloroform extraction and the upper phase after centrifugation was mixed with an equal volume of formamide gel loading dye to prepare samples for preparative electrophoresis (see below).
Purification of radiolabelled RNA oligonucleotides
ss17-A14 oligoribonucleotides with 5ʹ-hydroxyl/5ʹ-monophosphate, labelled at the 5ʹ-end with T4 PNK or at the 3ʹ-end with CutA or T4 RNA ligase I were purified following separation in 10% denaturing polyacrylamide gel containing 8 M urea. Labelled oligonucleotides were visualized by UV shadowing, slices of the gel were excised, crushed with pipette tip after freezing in liquid nitrogen and the RNAs were eluted overnight at room temperature with the use of buffer containing 100 mM Tris-HCl pH 8.0, 12.5 mM EDTA, 150 mM NaCl, and 1% SDS in the presence of an equal volume of acidic (pH 4.3) phenol:chloroform (1:1, v:v). Following centrifugation at 21000xg for 15 minutes at room temperature, RNA was precipitated from the supernatant using 1 volume of isopropanol and 1 µl of glycogen (20 mg/ml). Pellets were washed with 1 ml of 80% ethanol, air-dried and re-suspended in RNase-free water.
Electrophoretic analysis of GLuc and FLuc mRNAs
GLuc and FLuc transcripts 3ʹ-labelled using CutA and CTP*/UTP* or equivalent amounts of unlabelled GLuc/FLuc mRNAs were run in two sorts of 1% agarose gels: native in 1xTBE or denaturing (with 0.925% formaldehyde) in 1xNBC buffer (50 mM boric acid, 1 mM sodium acetate, 5 mM NaOH). Following electrophoretic separation in the native agarose gels stained with ethidium bromide (EtBr), RNA was immobilized on the nylon membrane (Whatman Nytran N; Cytiva) by overnight capillary transfer in 20xSSC (3 M NaCl, 0.3 M sodium citrate). RNA was fixed on membrane by ultraviolet cross-linking and visualized by staining with methylene blue solution (0.02% w/v in 0.3 M sodium acetate pH 5.2). Labelled mRNAs were visualized using Typhoon FLA 9000 scanner. For denaturing agarose-formaldehyde gel, the procedure was essentially the same, however neither the gel nor the samples contained EtBr. RiboRulerTM High Range RNA Ladder (Thermo Fisher Scientific) was used as a molecular weight marker during RNA electrophoresis in both kinds of agarose gels.
Northern blot detection of GLuc and FLuc transcripts
Probes for detection of GLuc or FLuc mRNAs were generated by amplification of CDS fragments in PCR with GLuc_qpcr_f1-GLuc_qpcr_r4 or FLuc_qpcr_f2-FLuc_qpcr_r1 (see Table S1 for nucleotide sequences) primer pairs (0.2 µM in each case) and pJET_GLuc or pJET_FLuc as templates, respectively, and using Phusion™ High-Fidelity DNA Polymerase (ThermoScientific) with 1xHF buffer, 0.2 mM dNTP Mix and 3% DMSO. The following PCR profile was utilized: initial denaturation at 98°C for 3 minutes, 35 cycles of amplification (with denaturation at 98°C, annealing at 55°C and elongation at 72°C carried out for 10, 30 and 45 seconds, respectively) and final elongation at 72°C for 5 minutes. Amplified products were separated in a preparative agarose gel stained with GelGreen and visualized using blue light transilluminator. Bands of the expected size were excised from the gel and purified using Gel-Out kit (A&A Biotechnology) according to manufacturer’s instructions. Approximately 10 ng of gel-purified DNA was labelled by random-priming with α-32P[dATP] and DecaLabel™ DNA Labelling Kit (Thermo Scientific) according to manufacturer’s instructions. Hybridizations with nylon membrane containing immobilized GLuc and FLuc transcripts transferred from denaturing agarose-formaldehyde gel (see above) were performed in PerfectHyb™ Plus hybridization buffer (Sigma-Aldrich) at 63°C. After each hybridization, membrane was washed with 2xSSC, 0.1% SDS at 63°C and exposed to a PhosphorImager screen (FujiFilm), which was scanned following exposure using a FLA 9000 scanner. Between hybridizations probe was stripped off the membrane at 65°C using boiling 0.1% SDS.
Exoribonuclease activity assays
Full-length S. cerevisiae Dis3 was purified as previously described [Citation22]. 3ʹ-5ʹ exoribonuclease assays were carried out in 25 μl reaction mixtures containing 10 mM Tris-HCl pH 8.0, 75 mM NaCl, 170 μM MgCl2, and 1 mM 2-mercaptoethanol, using 0.1 μM of protein and 2 μM of appropriate unlabelled RNA substrate supplemented with radiolabelled counterpart (approximately 100 cps of radioactivity per reaction, as estimated with handheld ionizing radiation survey instrument). Xrn1 5ʹ-3ʹ exoribonuclease assays were performed in an analogous manner, using commercial yeast enzyme (0.04 U/μl) and NEBuffer 3 (New England Biolabs). Reaction mixtures were incubated at 37°C for 0–60 minutes, and eventually stopped by addition of one volume of formamide gel loading dye. Reaction products were resolved either in denaturing 20% polyacrylamide/8 M urea/1x TBE gels or in 10% sequencing PAGE and visualized using Typhoon FLA 7000 PhosphorImager (FUJI).
Author contributions
RT conceived the studies, designed all experiments and performed all tests with synthetic RNA oligonucleotide substrates, analysed data and wrote the manuscript. KK participated in the development of CutA purification protocol and performed SEC-MALS analysis. KD carried out in vitro synthesis and CutA-mediated labelling of mRNA molecules. MH-S purified CutA variants with mutations. AD came up with an idea of possible use of CutA in RNA labelling procedure. All authors read and approved the final version of the manuscript.
Supplemental Material
Download PDF (12.8 MB)Acknowledgments
We acknowledge Dr Roman J. Szczesny, a Head of the Laboratory of RNA Biology, IBB PAS, for sharing most of the fluorescently labelled RNA oligonucleotide substrates used in this work. We thank Dr Pawel J. Sikorski from CENT UW for providing templates for mRNA in vitro synthesis and PCR primers for generation of northern blot probes. We also acknowledge an anonymous reviewer of our manuscript, whose valuable suggestions prompted us to perform labelling reactions with a broader spectrum of RNA substrates.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Liudkovska V, Dziembowski A. Functions and mechanisms of RNA tailing by metazoan terminal nucleotidyltransferases. Wiley Interdiscip Rev RNA. 2021;12(2):e1622.
- Yu S, Kim VN. A tale of non-canonical tails: gene regulation by post-transcriptional RNA tailing. Nat Rev Mol Cell Biol. 2020;21(9):542–556.
- Shukla A, Yan J, Pagano DJ, et al. Poly(UG)-tailed RNAs in genome protection and epigenetic inheritance. Nature. 2020;582(7811):283–288.
- Preston MA, Porter DF, Chen F, et al. Unbiased screen of RNA tailing activities reveals a poly(UG) polymerase. Nat Methods. 2019;16(5):437–445.
- Malik D, Kobylecki K, and Krawczyk P, et al. Structure and mechanism of CutA, RNA nucleotidyl transferase with an unusual preferece for cytosine. Nucleic Acids Res. 2020;48(16):9387–9405.
- Kobylecki K, Kuchta K, Dziembowski A, et al. Biochemical and structural bioinformatics studies of fungal CutA nucleotidyltransferases explain their unusual specificity toward CTP and increased tendency for cytidine incorporation at the 3ʹ-terminal positions of synthesized tails. RNA. 2017;23(12):1902–1926.
- Tomecki R, Drazkowska K, Krawczyk A, et al. Purification of eukaryotic exoribonucleases following heterologous expression in bacteria and analysis of their biochemical properties by in vitro enzymatic assays. Methods Mol Biol. 2015;1259:417–452.
- Wang Z, Kiledjian M. The poly(A)-binding protein and an mRNA stability protein jointly regulate an endoribonuclease activity. Mol Cell Biol. 2000;20(17):6334–6341.
- Schwarz DS, Tomari Y, Zamore PD. The RNA-induced silencing complex is a Mg2+-dependent endonuclease. Curr Biol. 2004;14(9):787–791.
- Lebreton A, Tomecki R, Dziembowski A, et al. Endonucleolytic RNA cleavage by a eukaryotic exosome. Nature. 2008;456(7224):993–996.
- Aphasizhev R, Simpson L. Isolation and characterization of a U-specific 3ʹ-5ʹ-exonuclease from mitochondria of Leishmania tarentolae. J Biol Chem. 2001;276(24):21280–21284.
- Clouet-d’Orval B, Rinaldi D, Quentin Y, et al. Euryarchaeal beta-CASP proteins with homology to bacterial RNase J have 5ʹ- to 3ʹ-exoribonuclease activity. J Biol Chem. 2010;285(23):17574–17583.
- Klocker N, Weissenboeck FP, Rentmeister A. Covalent labeling of nucleic acids. Chem Soc Rev. 2020;49(23):8749–8773.
- Rio DC. 5′-End Labeling of RNA with [γ-32P]ATP and T4 polynucleotide kinase. Cold Spring Harb Protoc. 2014;2014(4):441–443.
- Gaastra W, Josephsen J. Radiolabeling of DNA using polynucleotide kinase. Methods Mol Biol. 1985;2:263–268.
- Nilsen TW. 3′-End Labeling of RNA with [5′-32P]Cytidine 3′,5′-Bis(Phosphate) and T4 RNA Ligase 1. Cold Spring Harb Protoc. 2014;2014(4):444–446.
- England TE, Bruce AG, and Uhlenbeck OC. Specific labeling of 3ʹ termini of RNA with T4 RNA ligase. Methods Enzymol. 1980;65(1):65–74.
- England TE, Uhlenbeck OC. 3ʹ-terminal labelling of RNA with T4 RNA ligase. Nature. 1978;275(5680):560–561.
- Lingner J, Keller W. 3ʹ-end labeling of RNA with recombinant yeast poly(A) polymerase. Nucleic Acids Res. 1993;21(12):2917–2920.
- Rio DC. 3ʹ-end labeling of RNA with yeast poly(A) polymerase and 3ʹ-deoxyadenosine 5ʹ-[alpha-32P]triphosphate. Cold Spring Harb Protoc. 2014;2014(6):687–689.
- Huang Z, Szostak JW. A simple method for 3ʹ-labeling of RNA. Nucleic Acids Res. 1996;24(21):4360–4361.
- Lorentzen E, Basquin J, Tomecki R, et al. Structure of the active subunit of the yeast exosome core, Rrp44: diverse modes of substrate recruitment in the RNase II nuclease family. Mol Cell. 2008;29(6):717–728.
- Tomecki R, Kristiansen MS, Lykke-Andersen S, et al. The human core exosome interacts with differentially localized processive RNases: hDIS3 and hDIS3L. EMBO J. 2010;29(14):2342–2357.
- Zinder JC, Wasmuth EV, Lima CD. Nuclear RNA exosome at 3.1 Å reveals substrate specificities, RNA paths, and allosteric inhibition of Rrp44/Dis3. Mol Cell. 2016;64(4):734–745.
- Wu S, Mao G, Kirsebom LA. Inhibition of bacterial RNase P RNA by phenothiazine derivatives. Biomolecules. 2016;6(3):38.
- Jung E, Lee J, Hong HJ, et al. RNA recognition by a human antibody against brain cytoplasmic 200 RNA. RNA. 2014;20(6):805–814.
- Nikpour N, Salavati R. The RNA binding activity of the first identified trypanosome protein with Z-DNA-binding domains. Sci Rep. 2019;9(1):5904.
- Xing L, Niu M, Zhao X, et al. Roles of the linker region of RNA helicase A in HIV-1 RNA metabolism. PLoS One. 2013;8(11):e78596.
- Sikorski PJ, Warminski M, Kubacka D, et al. The identity and methylation status of the first transcribed nucleotide in eukaryotic mRNA 5ʹ cap modulates protein expression in living cells. Nucleic Acids Res. 2020;48(4):1607–1626.