
Abstract
Autophagy is a lysosomal degradation pathway of cellular components that displays antiinflammatory properties in macrophages. Macrophages are critically involved in chronic liver injury by releasing mediators that promote hepatocyte apoptosis, contribute to inflammatory cell recruitment and activation of hepatic fibrogenic cells. Here, we investigated whether macrophage autophagy may protect against chronic liver injury. Experiments were performed in mice with mutations in the autophagy gene Atg5 in the myeloid lineage (Atg5fl/fl LysM-Cre mice, referred to as atg5−/−) and their wild-type (Atg5fl/fl, referred to as WT) littermates. Liver fibrosis was induced by repeated intraperitoneal injection of carbon tetrachloride. In vitro studies were performed in cultures or co-cultures of peritoneal macrophages with hepatic myofibroblasts. As compared to WT littermates, atg5−/− mice exposed to chronic carbon tetrachloride administration displayed higher hepatic levels of IL1A and IL1B and enhanced inflammatory cell recruitment associated with exacerbated liver injury. In addition, atg5−/− mice were more susceptible to liver fibrosis, as shown by enhanced matrix and fibrogenic cell accumulation. Macrophages from atg5−/− mice secreted higher levels of reactive oxygen species (ROS)-induced IL1A and IL1B. Moreover, hepatic myofibroblasts exposed to the conditioned medium of macrophages from atg5−/− mice showed increased profibrogenic gene expression; this effect was blunted when neutralizing IL1A and IL1B in the conditioned medium of atg5−/− macrophages. Finally, administration of recombinant IL1RN (interleukin 1 receptor antagonist) to carbon tetrachloride-exposed atg5−/− mice blunted liver injury and fibrosis, identifying IL1A/B as central mediators in the deleterious effects of macrophage autophagy invalidation. These results uncover macrophage autophagy as a novel antiinflammatory pathway regulating liver fibrosis.
Abbreviations
| ATG | = | autophagy related |
| ACTA2 | = | actin, α 2, smooth muscle, aorta |
| CCL | = | chemokine (C-C motif) ligand |
| CCl4 | = | carbon tetrachloride |
| CCR2 | = | chemokine (C-C motif) receptor 2 |
| CM | = | conditioned medium |
| CXCL | = | chemokine (C-X-C motif) ligand |
| ADGRE1/EMR1 | = | adhesion G protein-couple receptor E1 |
| FBS | = | fetal bovine serum |
| GOT | = | glutamic-oxaloacetic transaminase |
| GPT | = | glutamic-pyruvate transaminase (alanine aminotransferase) |
| IL | = | interleukin |
| IL1RN | = | interleukin 1 receptor antagonist |
| LC3 | = | microtubule-associated protein 1 light chain 3 |
| LPS | = | lipopolysaccharide |
| MAPK | = | mitogen-activated protein kinase |
| MMP | = | matrix metallopeptidase |
| MO | = | mineral oil |
| MPO | = | myeloperoxidase |
| NAC | = | N-acetylcysteine |
| RELA | = | v-rel avian reticuloendotheliosis viral oncogene homolog A |
| ROS | = | reactive oxygen species |
| RT-PCR | = | real time polymerase chain reaction |
| TGFB1 | = | transforming growth factor, β 1 |
| TIMP1 | = | TIMP metallopeptidase inhibitor 1 |
| TNF | = | tumor necrosis factor |
| TUNEL | = | terminal deoxynucleotidyl transferase dUTP nick end labeling |
| WT | = | wild type. |
Introduction
Liver fibrosis is a common pathological consequence of chronic liver diseases, and its endstage, cirrhosis, represents a major cause of morbidity and mortality worldwide, owing to life-threatening complications of portal hypertension and liver failure and to the high risk of incident hepatocellular carcinoma. The development of fibrosis results from a sustained wound-healing process in response to chronic liver injury, characterized by increased production of matrix proteins and decreased matrix remodeling.Citation1 Experimental models of liver fibrosis highlighted that macrophages exert a dual function in this process.Citation2 Thus, hepatic macrophages consisting of resident Kupffer cells and infiltrating monocytes are critically involved in the initiation and perpetuation of fibrosis by releasing mediators that promote hepatocyte apoptosis, contribute to inflammatory cell recruitment and activate hepatic myofibroblasts.Citation3 In addition to their role in fibrosis pro-gression, distinct subsets of monocyte/macrophages display antifibrogenic properties because of their antiinflammatory propertiesCitation4 and of their matrix-degrading role during fibrosis regression.Citation2,5
Macroautophagy (hereafter referred to as autophagy) is an evolutionarily conserved process carrying out degradation of cytoplasmic constituents dedicated to the turnover of cellular components.Citation6 This process involves the coordinated action of autophagy-related (ATG) proteins, which induce the formation of a double-membraned vesicle, the autophagosome, that subsequently fuses with a lysosome to form an autolysosome. This maturation leads to the degradation of sequestered cytoplasmic components by acid proteases and hydrolases contained in the autolysosome. Autophagy occurs constitutively at basal rates in virtually all cells to clear damaged organelles and insoluble protein aggregates, thus regulating cellular homeostasis and cell death/survival.Citation6,7 Recent studies have identified autophagy as a major regulator of both the innate and adaptive immune responses such as clearance of intracellular pathogens, antigen presentation to MHC class II, thymic selection and lymphocyte differenciation.Citation8 More recently, beneficial roles for autophagy in macrophages have been described in efferocytosis, protection against oxidative stress and excessive production of inflammatory cytokines, in particular IL1A and IL1B.Citation9-15 In keeping with these data, macrophage autophagy has emerged as an interesting therapeutic target in the context of a variety of inflammatory diseases, including colitis, atherosclerosis, and tuberculosis.Citation9-12
In the current study, we explored the role of macrophage autophagy in chronic liver injury, by using the Cre-loxP system to specifically knock down the autophagy gene Atg5 in the myeloid lineage. We demonstrate that macrophage autophagy limits IL1A and IL1B secretion, with resulting protective effects against liver injury, inflammatory cell recruitment, and fibrogenesis.
Results
Macrophages from atg5−/− mice are resistant to autophagy and more susceptible to LPS-induced IL1A/B release
We generated myeloid cell-specific Atg5 knockout (Atg5fl/fl LysM-Cre or atg5−/−) mice by crossing Atg5flox/flox mice with transgenic mice expressing the Cre recombinase under the control of the lysozyme M promoter (LysMCre+/+ mice). Autophagy dysfunction was confirmed in Kupffer cells isolated from atg5−/− mice that were treated with the autophagy inducer rapamycin. Autophagy induction is associated with the conversion of the cytosolic form of MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3 ; LC3-I) to the phagophore- and autophagosome-bound form of LC3 (LC3-II), which can be detected as a punctate signal by immunocytochemistry.Citation16 Moreover, the expression of SQSTM1/p62 protein is inversely correlated with autophagy level.Citation16 As exptected, rapamycin increased the number of LC3 puncta and decreased the number of SQSTM1/p62 puncta in WT cells but not in atg5−/− Kuppfer cells ().
Figure 1 (See previous page). Macrophage from atg5−/− mice show impaired autophagy and proinflammatory properties. (A) LC3 and SQSTM1 protein expression and quantification by immunocytochemistry in Kuppfer cells exposed for 6 h to 100 nM rapamycin or its vehicle. Data are shown as mean ± SEM of 4 independent experiments. *, p < 0.05 for rapamycin vs vehicle, &, p < 0.05 for WT vs atg5−/−. (B) ELISA analysis of IL1A and IL1B in supernatant fractions from Kupffer cells and neutrophils isolated from atg5−/− or WT mice and exposed to 10 ng/ml LPS for 6 h. Data are shown as mean ± SEM and are representative of 2 independent experiments. &, p < 0.05 for WT vs atg5−/−. (C) Left, Il1a and pro-il1b mRNA expression in peritoneal macrophages isolated from atg5−/− or WT mice and exposed to 10 ng/ml LPS for 6 h. Right, ELISA analysis of IL1A and IL1B in supernatant fractions from peritoneal macrophages isolated from atg5−/− or WT mice and exposed to 10 ng/ml LPS for 24 h. Data are shown as mean ± SEM and are representative of 2 independent experiments. &, p < 0.05 for WT vs atg5−/−. (D) RT-PCR analysis of Il1a and pro-il1b mRNA in RAW264.7 cells exposed for 1 h to 100 nM rapamycin or vehicle, and further incubated with 10 ng/ml LPS for 6 h. Data are the mean ± SEM from sextuplate repeats. *, p < 0.05 for vehicle vs rapamycin.
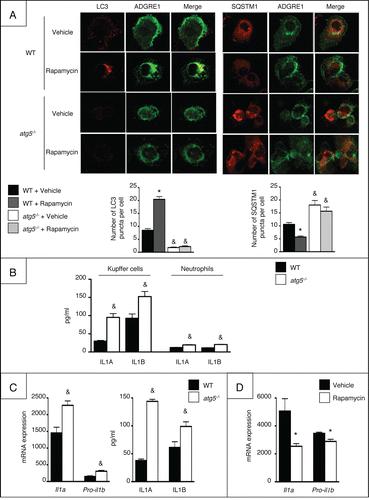
Recent studies have shown that loss of macrophage autophagy is associated with enhanced IL1A and IL1B secretion.Citation9-15 In keeping with this finding, Kupffer cells isolated from atg5−/− mice exposed to LPS exhibited increased secretion of IL1A and IL1B, as compared to Kupffer cells from WT counterparts (). Similar results were obtained using peritoneal macrophages both at the mRNA and protein levels (). Conversely, rapamycin reduced the expression of Il1a and pro-il1b in LPS-exposed macrophages (). Finally, secretion of IL1A and IL1B was hardly detectable in neutrophils isolated from atg5−/− mice and exposed to LPS (), as compared to levels in macrophages, suggesting that neutrophils marginally contribute to IL1 production after Atg5 deletion in the myeloid lineage.
We next investigated the mechanisms of the enhanced production of IL1A and IL1B by Atg5-deleted macrophages and first focused on ROS, since ROS play an important role in the regulation of IL1A and IL1B gene expression and signaling. atg5−/− macrophages displayed higher ROS production in response to LPS than WT counterparts (). Moreover, Il1a and pro-il1b mRNA expression and secretion were blunted by N-acetylcysteine (NAC), demonstrating that Atg5 mutation enhanced IL1A/B secretion via ROS generation (). We next dissected the signaling pathways regulating LPS-induced IL1A and IL1B secretion. atg5−/− macrophages showed enhanced MAPK14 activation as compared to WT cells, as shown by detection of the phosphorylated MAPK14 isoform by immunocytochemistry and western blotting (). In contrast, phosphorylation of RELA and MAPK8 were not modified (not shown). Moreover, MAPK14 inhibition by SB203580 reduced the expression of Il1a and pro-il1b in atg5−/− macrophages, indicating that enhanced activation of MAPK14 led to increased production of IL1A and IL1B in Atg5-deleted macrophages ().
Figure 2. ROS generation and activation of MAPK14 is associated with enhanced IL1A/B production by macrophages from atg5−/− mice. (A) ROS production in peritoneal macrophages isolated from atg5−/− or WT mice and exposed to 10 ng/ml LPS. Data are the mean ± SEM (n = 4). &, p < 0.05 for WT vs atg5−/− (B) Il1a and pro-il1b mRNA expression and production in peritoneal macrophages isolated from atg5−/− or WT mice and exposed to 10 mM NAC or its vehicle for 1 h and further stimulated with 10 ng/ml LPS for 6 h. Data are the mean ± SEM from sextuplate repeats. *, p < 0.05 for NAC vs vehicle and &, p < 0.05 for WT vs atg5−/−. (C) Representative P-MAPK14 protein expression by immunocytochemistry in peritoneal macrophages exposed for 15, 30 or 60 min to 10 ng/ml LPS or its vehicle. (D) P-MAPK14 and MAPK14 expression by western blotting in peritoneal macrophages exposed for 15 or 30 min to 10 ng/ml LPS or its vehicle. *, p < 0.05 for WT vs atg5−/− (E) Il1a and pro-il1b mRNA expression in peritoneal macrophages isolated from atg5−/− or WT mice and exposed to 10 μM SB203580 for 1 h and further stimulated with 10 ng/ml LPS for 6 h. Data are the mean ± SEM from sextuplate repeats. *, p < 0.05 for vehicle vs SB203580, &, p < 0.05 for WT vs atg5−/−.
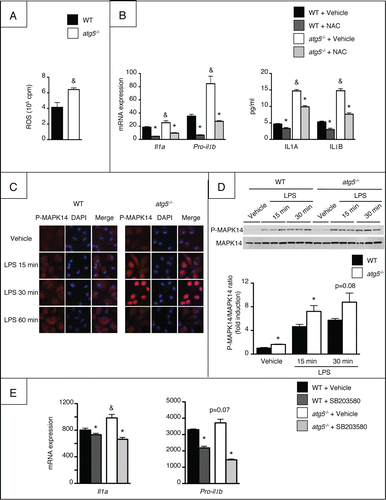
Together, these data demonstrate that Atg5-deficient macrophages are resistant to autophagy, and acquire a more severe proinflammatory phenotype.
Inflammatory cell recruitement and hepatic inflammation are enhanced in atg5−/− mice after CCl4-induced liver damage
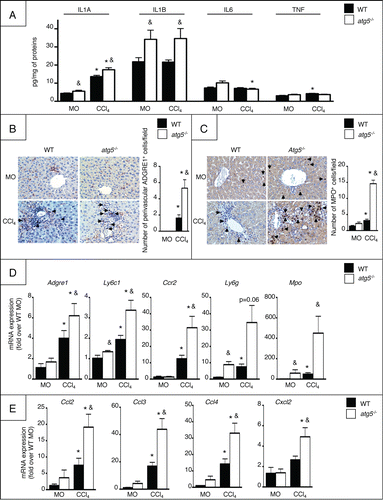
We next evaluated the consequences of Atg5 deletion in myeloid cells on chronic liver injury in mice exposed to repeated injections of CCl4.Citation17 In this model, repeated hepatocyte damage elicited by CCl4 is associated with activation of Kupffer cells and infiltration of the liver by inflammatory cells, resulting in an exagerated wound healing response and abnormal matrix remodelling driven by hepatic myofibroblasts. Quantification of the levels of proinflammatory cytokines in the liver of vehicle (MO)- and CCl4-exposed atg5−/− mice showed higher levels of IL1A in both conditions when compared to WT littermates (). Moreover, the hepatic levels of IL1B were increased in MO-treated atg5−/− mice but not further enhanced in CCl4-exposed animals (). In contrast, the hepatic levels of IL6 and TNF were similar in both groups of animals (). Livers from CCl4-exposed atg5−/− mice showed a higher number of recruited monocytes () and neutrophils () compared to WT mice, as shown by ADGRE1/EMR1 and MPO (myeloperoxidase) immunostaining, respectively. This increased leucocyte infiltration was associated with higher hepatic mRNA expression of macrophage/monocyte markers (Adgre1, Ly6c1, and Ccr2,), and of neutrophil genes (Ly6g and Mpo,). However, the expression of Adgre1 and Ly6c1 was not modified in atg5−/− macrophages, suggesting that the higher hepatic expression of these markers might be related to increased monocyte infiltration in CCl4-exposed atg5−/− cells, rather than to an increased expression of these markers per macrophage (Fig. S1). Moreover, the hepatic mRNA levels of a number of chemokines essential for monocyte and neutrophil recruitment (Ccl2, Ccl3, Ccl4, and Cxcl2) were significantly higher in these mice, both at basal levels and following CCl4 exposure (). These data indicate that autophagy deficiency in the myeloid lineage enhances inflammatory cell recruitment and hepatic inflammation.
Figure 3. atg5−/− mice show enhanced liver inflammation. (A) ELISA quantification of cytokine levels in liver homogenates in WT or atg5−/− mice exposed to CCl4 for 2.5 weeks or its vehicle (mineral oil, MO). Representative ADGRE1 (B) and MPO (C) staining (original magnification x400) and quantification in WT or atg5−/− mice exposed to CCl4 for 2.5 wk or its vehicle (MO). Arrows indicate positive cells. (D) Hepatic mRNA expression of Adgre1, Ly6c1, Ccr2, Ly6g, and Mpo in WT or atg5−/− mice exposed to CCl4 for 2.5 wk or its vehicle (MO). (E) Hepatic mRNA expression of Ccl2, Ccl3, Ccl4 and Cxcl2 in WT or atg5−/− mice exposed to CCl4 for 2.5 wk or its vehicle (MO). Data are shown as mean ± SEM; *, p < 0.05 for MO vs CCl4 and &, p < 0.05 for WT vs atg5−/−. n = 3 for WT mice MO; n = 4 for atg5−/− mice MO; n = 14 for WT mice CCl4; n = 12 for atg5−/− mice CCl4.
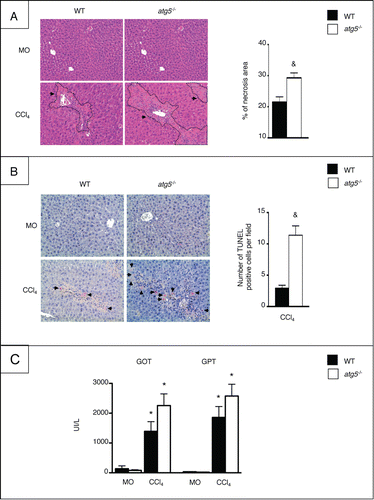
Hepatocyte apoptosis is worsened in atg5−/− mice after CCl4-induced liver damage
We next investigated liver damage in atg5−/− mice exposed to CCl4 by studying the extent of hepatocyte necrosis and apotosis. Analysis of liver histology in hematoxylin and eosin-stained liver sections showed that hepatocyte death was more pronounced in atg5−/− mice than in WT counterparts, following CCl4 exposure (). Moreover, the number of TUNEL-positive hepatocytes was significantly higher in CCl4-exposed atg5−/− mice than in WT animals (). Finally, serum GOT (glutamic-oxaloacetic transaminase) and GPT (glutamic-oxaloacetic transaminase [aspartate aminotransferase]) levels were markedly elevated in atg5−/− mice exposed to CCl4, although the difference with WT mice did not reach statistical significance (). Differences between CCl4-treated atg5−/− and WT mice were not due to an impact of Atg5 deletion on CCl4 metabolism, because the 2 groups showed no difference in the expression of CYP2E1 (cytochrome P450, family 2, subfamily E, polypeptide 1; not shown), a cytochrome isoform that metabolizes CCl4 into hepatotoxic metabolites. Taken together, these data indicate that autophagy inactivation in the myeloid lineage enhances liver injury.
Figure 4. atg5−/− mice display enhanced liver injury. (A) Left, representative staining of liver tissue sections stained with hematoxylin and eosin (original magnification x200) in WT or atg5−/− mice exposed to CCl4 for 2.5 wk or its vehicle (MO). Arrows indicate necrotic areas. Right, quantification of necrosis area by morphometry. (B) Left, representative TUNEL staining (original magnification x200) in WT or atg5−/− mice exposed to CCl4 for 2.5 wk or its vehicle (MO). Arrows indicate TUNEL-positive cells. Right, quantification of TUNEL staining expressed as number of positive cells per field. (C) Serum levels of GOT and GPT in WT or atg5−/− mice exposed to CCl4 for 2.5 wk or MO. Data are shown as mean ±SEM ; *, p < 0.05 for MO vs CCl4 and &, p<0.05 for WT vs atg5−/−. n = 3 for WT mice MO; n = 4 for atg5−/− mice MO; n = 14 for WT mice CCl4; n = 12 for atg5−/− mice CCl4.
Liver fibrosis is exacerbated in atg5−/− mice after chronic CCl4 treatment
We next investigated the role of macrophage autophagy in liver fibrosis in mice exposed to CCl4 for 2.5 or 4 wk. atg5−/− mice developed significantly more fibrosis than WT animals, as evidenced by increased fibrosis area quantified in liver tissue sections stained with Sirius Red (). In addition, immunohistochemical detection of ACTA2 (actin, α 2, smooth muscle, aorta) showed a higher number of fibrogenic cells in these animals (). Finally, the hepatic mRNA expression of the fibrogenic genes Tgfb1, Mmp9, and Serpine1 was enhanced in atg5−/− mice exposed to CCl4 (). These results demonstrate that atg5−/− mice show exacerbated hepatic fibrosis following exposition to CCl4.
Figure 5. atg5−/− mice display exacerbated liver fibrosis. (A) Left, representative liver tissue sections stained with Sirius Red (original magnification x200) in mice exposed to CCl4 for 2.5 or 4 wk. Right, quantification of fibrosis area by morphometry. (B) Left, representative hepatic ACTA2 immunostaining (original magnification x200) in mice exposed to CCl4 for 2.5 or 4 wk. Right, quantification of ACTA2 immunostaining by morphometry. (C) RT-PCR analysis of hepatic Tgfb1, Mmp9, and Serpine1 mRNA expression in mice exposed to CCl4 for 2.5 wk. Data are shown as mean ± SEM; *, p < 0.05 for MO vs CCl4 and, & p < 0.05 for WT vs atg5−/−. n = 3 for WT mice MO 2.5 wk; n = 5 for WT mice MO 4 wk; n = 4 for atg5−/− mice MO 2.5 wk; n = 5 for atg5−/− mice MO 4 wk; n = 14 for WT mice CCl4 2.5 wk; n = 14 for WT mice CCl4 4 wk; n = 12 for atg5−/− mice CCl4 2.5 wk; n = 11 for atg5−/− mice CCl4 4 wk.
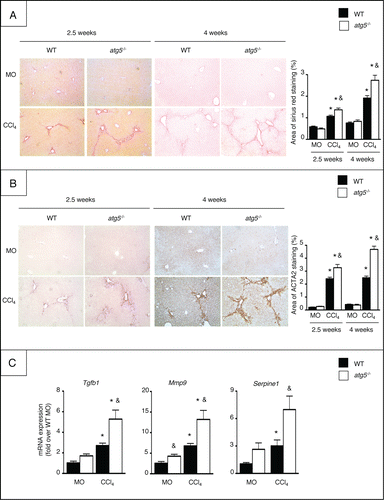
Autophagy-deficient macrophages enhance the fibrogenic properties of hepatic myofibroblasts via an IL1-dependent pathway
We further evaluated the impact of macrophage autophagy on the profibrogenic properties of hepatic myofibroblasts in conditioned medium experiments. Mouse hepatic myofibroblasts were cultured in the presence of conditioned medium (CM) collected from LPS-exposed peritoneal macrophages isolated from atg5−/− or WT mice. The mRNA expression of Timp1, Serpine1, and Mmp9 was significantly enhanced in myofibroblasts exposed to CM collected from atg5−/− cells (). Anti-IL1A/B neutralizing antibodies added to atg5−/− CM totally prevented the increase in profibrogenic genes in hepatic myofibroblasts (), whereas, conversely, direct exposure of the cells to IL1A and IL1B resulted in the induction of these genes ().
Figure 6. Atg5-deficient macrophages enhance the fibrogenic properties of hepatic myofibroblasts, via an IL1-dependent pathway. (A) Conditioned media (CM) were obtained from atg5-/− or WT peritoneal macrophages exposed for 24 h to LPS, and were further incubated for 1 h with IL1A/B neutralizing antibodies or control IgG. RT-PCR analysis of Timp1, Serpine1 and Mmp9 mRNA was performed in myofibroblasts exposed for 6 h to CM. *, p<0.05 for CM atg5−/− + control IgG vs CM WT + control IgG and &, p<0.05 for CM atg5−/− + control IgG vs CM atg5−/− + anti-IL1A/B. Data are shown as mean ± SEM and are representative of 2 independent experiments. (B) RT-PCR analysis of Timp1, Serpine1 and Mmp9 mRNA in hepatic myofibroblasts exposed to recombinant IL1A or IL1B for 6 h. *, p<0.05 for vehicle vs IL1. Data are the mean ± SEM of 2 independent experiments.
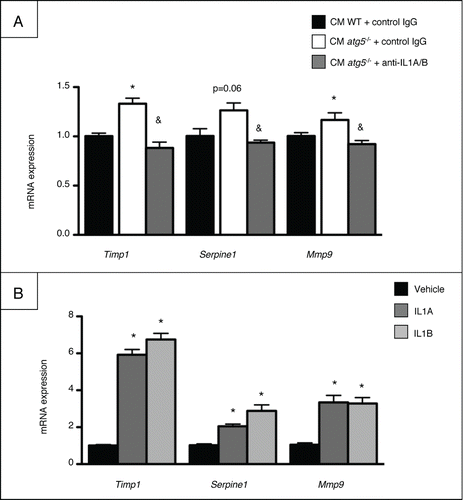
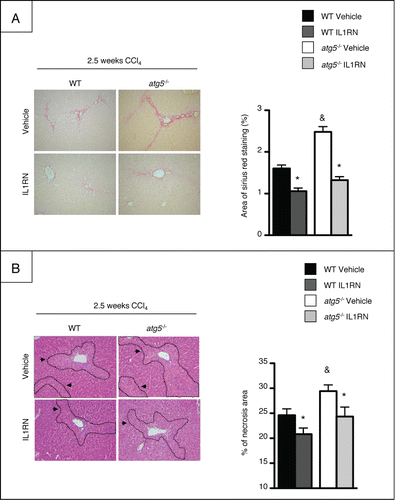
Treatment with recombinant IL1RN rescues atg5−/− mice from CCl4-induced liver fibrosis and injury
Blockade of IL1A/B signaling was performed with IL1RN in order to provide a direct link between macrophage Atg5 deficiency, deregulated IL1A/B production, and liver damage. Administration of recombinant IL1RN to CCl4-exposed atg5−/− mice reduced the Sirius Red staining area to levels comparable to that of WT mice (). Moreover, recombinant IL1RN caused a small but significant amelioration of CCl4-induced liver injury () as shown by reduced necrosis area in atg5−/− mice. These data demonstrate that macrophage autophagy protects against liver fibrosis and injury by limiting IL1A and B release.
Figure 7. Treatment with recombinant IL1RN rescues atg5−/− mice from CCl4-induced liver fibrosis and injury. Mice were exposed to CCl4 and treated with recombinant IL1RN or its vehicle for 2.5 wk (A) Left, representative liver tissue sections stained with Sirius Red (original magnification x200). Right, quantification of fibrosis area by morphometry. (B) Left, representative staining of liver tissue sections stained with hematoxylin and eosin (original magnification x200). Arrows indicate necrotic areas. Right, quantification of necrosis area by morphometry. *, p < 0.05 for IL1RN vs vehicle and &, p < 0.05 for atg5−/− vs WT. n = 7 for WT vehicle, n = 6 for atg5−/− vehicle, n = 4 for WT IL1RN and n = 5 for atg5−/− IL1RN.
Discussion
Hepatic macrophages are critically involved in the progression of chronic liver injury, and represent attractive targets for antifibrotic therapies. Therefore, identification of specific antiinflammatory pathways in these cells may serve as the basis for the design of novel antifibrotic strategies. In the present study, we uncover autophagy in macrophages as a novel antiinflammatory pathway controlling liver fibrosis.
Accumulating studies have highlighted the fact that the autophagic pathway in macrophages is a negative regulator of inflammation, by limiting inflammatory cytokine release from macrophages and reducing inflammatory cell recruitm-ent.Citation9-15 In line with these reports, we show that genetic inactivation of the autophagic gene Atg5 in myeloid cells leads to enhanced secretion of IL1A and IL1B both in isolated Kupffer cells and in the liver of mice exposed to repeated administration of carbon tetrachloride. Conversely, activation of autophagy by rapamycin limits IL1A and IL1B production by macrophages. Interestingly, our data demonstrate that macrophage autophagy regulates IL1B expression both at the mRNA and protein level, while another study reported that autophagy selectively affects IL1B production.Citation9 This discrepancy may result from differences in experimental design, in particular the duration of cell exposure to LPS. In line with previous findings,Citation10-14 we also demonstrate that autophagy-deficient macrophages show exacerbated production of reactive oxygen species and enhanced activation of MAPK14, that account for enhanced secretion of IL1A and IL1B in these cells.
A major feature of chronic liver injury resides in the influx of inflammatory cells into the damaged liver, that contributes to perpetuate liver injury and activation of fibrogenic cells.Citation1 We show that autophagy tightly limits accumulation of neutrophils and infiltration of monocytes into the injured liver. Interestingly, autophagy-mediated reduction of IL1A and IL1B secretion by macrophages may be involved, since IL1R-deficient mice show reduced neutrophilic infiltrate and liver injury, as well as resistance to liver fibrosis.Citation18 Whether limitation of IL1A and IL1B secretion is coordinated or sequential remains to be further explored, in light of a recent study showing that IL1A initiates sterile inflammation by promoting neutrophil recruitment, whereas IL1B rather perpetuates the inflammatory response by promoting the recruitment of macrophages.Citation19
A major point of the present study resides in the identification of macrophage autophagy as a novel antifibrogenic pathway. Indeed, our results demonstrate that mice specifically deleted for the autophagic gene Atg5 in myeloid cells are more susceptible to liver fibrosis elicited by chronic carbon tetrachloride administration. Consistent with in vivo data, our in vitro studies demonstrate that hepatic myofibroblasts exposed to conditioned media from autophagy-deficient macrophages show enhanced expression of fibrosis-related genes as compared to WT counterparts. In addition, anti-IL1A/B neutralizing antibobies blunt the profibrogenic effects of autophagy-deficient macrophage conditoned media. Finally, IL1RN administration to atg5−/− mice reduced liver fibrosis. These data identify IL1A/B as central mediators in the fibrogenic effects of macrophage autophagy invalidation. However, IL1B production is similarly enhanced in vehicle and CCl4-treated atg5−/− mice, suggesting a prominent role for IL1A in this effect. Compelling evidence demonstrates that IL1A/B are crucial mediators of liver fibrosis, as shown by the resistance of both IL1A- and IL1B-deficient mice to fibrosis in experimental models of alcohol- or diet-induced steatohepatitis.Citation20-22 Our results further shed light on macrophage autophagy as a novel paracrine pathway that controls IL1-dependent activation of hepatic myofibroblasts.
An additional mechanism by which macrophage autophagy may limit fibrosis progression is by limiting hepatocellular injury. Hepatocyte apoptosis is a common feature of chronic liver disease that contributes to the profibrogenic process by promoting survival of hepatic myofibroblasts and enhancing their profibrogenic properties.Citation23 In keeping with this concept, hepatoprotective strategies have shown encouraging antifibrogenic effects.Citation24 Recent data have shown that autophagy in hepatocytes limits apoptosis and protects against liver injury.Citation25-28 Our data, showing enhanced hepatocyte apoptosis in atg5−/− mice exposed to CCl4, highlight macrophage autophagy as an additional hepatoprotective mechanism that may contribute to its antifibrogenic effects. Increased hepatocyte death results from enhanced ILA/B secretion, because treatment of atg5−/− mice with IL1RN ameliorates liver damage. In addition, hepatocyte apoptosis could originate from enhanced accumulation of neutrophils observed in atg5−/− mice. Indeed, the prominent role of neutrophils has been demonstrated in different models of liver injury, including chronic plus binge ethanol feeding,Citation29 ischemia/reperfusion injury,Citation30 and acetamoniphen-Citation31 or concanavalin A-induced liver injuryCitation32 via the release of oxidative stress and cytotoxic mediators. Our data further extend autophagy as a hepatoprotective mechanism, not only operative in hepatocytes as a protection against cell death,Citation25-28 but also active in macrophages, with resulting antiapoptotic effects on hepatocytes via paracrine interactions. Autophagy has generally been considered as a protective pathway against chronic liver injury, because of the beneficial effects of hepatocyte autophagy on steatosis, apoptotis, and fibrosis.Citation25-28,33,34 Our data further argue for a beneficial effect of autophagy in macrophages, that prevents the excessive release of inflammatory cytokines. However, a complex scheme is emerging because autophagy in fibrogenic cells has emerged as a profibrogenic pathway that promotes upregulation of their fibrogenic properties.Citation35,36 Therefore, because autophagy elicits divergent and cell-specific effects during chronic liver injury, cell-specific delivery of drugs that exploit autophagic pathways is a prerequisite to further consider autophagy as a potential target for antifibrotic therapy.
In conclusion, our study reveals that specific disruption of autophagy in myeloid cells exacerbates liver fibrosis, hepatocellular injury and inflammation, by exacerbating IL1A/B production. These results identify macrophage autophagy as a novel protective pathway during chronic liver injury. Whether this pathway is also relevant to IL1-driven inflammation in other conditions such as fatty liver diseasesCitation21,37 remains to be investigated.
Materials and Methods
Animals and experimental design
Atg5flox/flox mice were kindly provided by Dr Noburo Mizushima (Japan).Citation38 Myeloid cell-specific Atg5 deficient (Atg5flox/flox LysCre+/−, Atg5fl/fl LysM-Cre) mice were generated by crossing Atg5flox/flox mice with lysozyme M-promoter Cre transgenic mice (Charles River, France). Atg5flox/flox LysMCre−/− mice were used as littermate WT controls. Animals were housed in pathogen-free animal facility and fed ad libitum. All animal procedures were approved by the Committee for the Care and Use of Laboratory Animals of the Paris-Est Creteil University (ComEth, Authorization N°12–082). Eight- to 14-wk-old mice were used. Liver fibrosis was induced by intraperitoneal injections of CCl4 (Sigma, 87031; 0.5 ml/kg body weight, 1:10 dilution in mineral oil [MO; Sigma, M5310]), twice a wk for up to 2.5 or 4 wk. Control animals received MO. Mice were starved overnight and sacrified 48 h after the last CCl4 injection. Mice were randomized into an MO-treated group (n=3 for WT 2.5 wk, n = 4 for atg5−/− 2.5 wk, n = 5 for WT 4 weeks, n = 5 for atg5−/− 4 wk) and CCl4-treated group (n = 14 for WT 2.5 wk, n = 12 for atg5−/− 2.5 weeks, n = 14 for WT 4 wk, n = 11 for atg5−/− 4 wk). When indicated, mice were treated with recombinant IL1RN (Merck, 407616; 25 mg/kg, ip) or its vehicle every d for 2.5 wk. Mice were randomized into a vehicle-treated group (n = 7 for WT and n = 6 for atg5−/−) and IL1RN-treated group (n = 4 for WT and n = 5 for atg5−/−).
Serum analysis
Blood was collected at the time of sacrifice. GOT (glutamic-oxaloacetic transaminase [aspartate aminotransferase]) and GPT (glutamic-pyruvate transaminase [alanine aminotransferase]) activities were measured on an automated analyzer in the Biochemistry Department of Henri Mondor Hospital.
Histological analysis
Hematoxylin and eosin and Sirius Red staining were performed on 4-μm thick formalin-fixed paraffin-embedded tissue sections. Sirius Red-stained areas from 10 fields (magnification x200) from 3 to 7 mice/group were quantified with ImageJ. Necrosis area from 5 to 9 fields (magnification x100) from 3 to 14 mice/group were quantified with ImageJ.
Cell culture
Kuppfer cells
Kuppfer cells were isolated from WT and atg5−/− mice after perfusion with liberase (Roche, 5 401 119 001) and differential centrifugation in Percoll (GE Healthcare, 17–0891–02) as previously described.Citation39 When indicated, adherent Kupffer cells were treated with 100 nM rapamycin (Sigma, R8781) or vehicle for 6 h. Cells used to detect LC3 were treated with 10 μm chloroquine (Sigma, C6628). When indicated, cells were treated with 10 mg/ml LPS (Sigma, L2887) or vehicle for 6 h.
Peritoneal macrophages
Peritoneal macrophages were harvested by lavage with phosphate-buffered saline (PBS; Life Technologies, 14200–067), 3 d after injection of 1.5 ml of sterile 4% thioglycolate medium (BD, 211716) into the peritoneal cavity of the mice. After red blood cells lysis with Red Blood Cell Lysis solution (Miltenyi Biotec, 130–094–183), cells were seeded in RPMI supplemented with 10% fetal bovine serum (FBS) and washed after 3 h of adhesion. Cells were then stimulated with 10 ng/ml LPS (Sigma, L2887) or vehicle for 6 h for mRNA expression or for 15, 30, or 60 min for immunocytochemical detection of phosphorylated (P)-MAPK14. When indicated, cells were treated with 10 mM NAC (Sigma, A9165) or 10 μM SB203580 (Sigma, S8307) for 1 h and further stimulated with 10 ng/ml LPS for 6 h.
Neutrophils
Mouse neutrophils were isolated from bone marrow originating from the femur and the tebia. Briefly, bone marrow cells were flushed with PBS containing 0.1% BSA (Sigma, A7030) and 5 mM EDTA. Following centrifugation at 600 g for 10 min at 4°C, pelleted cells were resuspended in 3 ml of 45% Percoll and neutrophils were purified over a 3-layer Percoll gradient (81%, 50% and 62%) by centrifugation at 1600 g for 30 min. Neutrophils were collected between the 81% and 62% Percol layers and washed in PBS.
RAW264.7 macrophages
Cells were seeded in DMEM supplemented with 10% FBS. After 24 h, cells were treated with 100 nM rapamycin or vehicle for 1 h and further incubated with 10 ng/ml LPS for 6 h.
Myofibroblasts
Mouse myofibroblasts were isolated after liberase (Roche, 5 401 119 001) perfusion of the liver and density gradient purification in Nycodenz (AbCys, 1002424), as previously described.Citation40 Cells between the fourth and the ninth passages were used. Cells were seeded in DMEM supplemented with 10% FBS, serum-starved for 24 h, and exposed to recombinant mouse IL1A (10 ng/ml, R&D systems, 400-ML-025), IL1B (10 ng/ml, Peprotech, 211–11B), or vehicle (0.1% BSA in H2O) for 6 h. Conditioned medium experiments were performed by adding for 6 h centrifuged conditioned medium obtained from LPS-stimulated peritoneal cells isolated from WT or atg5−/− mice. For neutralizing IL1A/B experiments, conditioned media were incubated for 1 h with polyclonal anti-IL1A (R&D Systems, AF-400NA) and anti-IL1B (R&D Systems, AF-401NA) antibodies or polyclonal IgG control antibody (R&D Systems, AB-108-C), according to the manufacturer's instructions.
Immunohistochemistry
Immunohistochemical detection of ACTA2 was carried out on paraffin-embedded liver tissue sections (4 μm) using the MOM immunodetection kit (Vector, PK2002) and a mouse monoclonal anti-ACTA2 antibody (1:1000; Sigma, 2547) according to the manufacturer's instructions. ACTA2-positive area from 10 fields (magnification x100) from 3 to 7 mice/group were quantified with ImageJ. No staining was observed when the primary antibody was omitted. Immunohistochemical detection of ADGRE1/EMR1 (Serotec, MCA497G) and MPO (Dako, A0398) was performed as previously described.Citation41 The number of ADGRE1- or MPO-positive cells from 5 to 9 fields (magnification x200) from 3 to 7 mice/group were quantified.
Immunocytochemistry
Cells were fixed in methanol, incubated in a blocking buffer containing 1% BSA and 0.2% Triton X-100 (Sigma, T8787), followed by incubation with an anti-LC3 antibody (1:200, clone 5F10; Nanotools, 0231–100), an anti-SQSTM1/p62 antibody (1:100; ProGen, GP62-C), an anti-ADGRE1 antibody (Serotec, MCA497G) or an anti-P-MAPK14 antibody (1:50; Cell Signaling Technology, 4631). Labeling was achieved using secondary antibodies (goat anti-mouse Alexa Fluor 555 (Life Technologies, A21424), goat anti-guinea pig Alexa Fluor 555 (Life Technologies, A21435), anti-rat-FITC (Serotec, STAR80F) or goat anti-rabbit Alexa Fluor 555 (Life Technologies, A31629)). Fluorescence was imaged on a Zeiss LSM-510 multitracking laser-scanning confocal microscope with a Helium/Neon laser at 543 nm and using AxioVision software (Carl Zeiss). No staining was observed when omitting the primary antibody. The number of LC3 or SQSTM1/p62 puncta per cell was quantified from 13 to 20 fields/condition.
TUNEL assay
Terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick-end labeling (TUNEL) staining was performed on paraffin-embedded tissue sections, using the In Situ Cell Death Detection Kit, POD (Roche, 11–684–809–910). TUNEL-positive cells from 10 fields (magnification ×200)/animal were quantified with ImageJ. Results are expressed as number of TUNEL-positive cells per field and were quantified from 6 animals/group.
Western blot analysis
Western blot analysis was performed with the following antibodies: rabbit anti-P-MAPK14 (Cell Signaling Technology, 9211) and rabbit anti-MAPK14 (Cell Signaling Technology, 9212) followed by incubation with a donkey anti-rabbit HRP-conjugated secondary antibody (GE Healthcare, NA934V). Proteins were visualized by an enhanced chemiluminescence assay kit (ECL Plus; GE Healthcare) using an Image Quant Las 4000 miniscanner (GE Healthcare) and signals were quantified using Image Quant software (GE Healthcare).
Immunoassays
Cytokines were quantified by Enzyme Linked Immunosorbant Assay (ELISA, R&D Systems, M6000B for IL6, MTA00 for TNF, MLA00 for IL1A and MMLB00C for IL1B) on liver homogenates and culture supernatant fractions from peritoneal macrophages according to the manufacturer's instructions. Cytokine levels were normalized per mg of protein.
RNA preparation and real-time polymerase chain reaction
Total RNA was extracted using the RNeasy Mini kit (Qiagen, 74106). cDNA was synthesized using the High Capacity cDNA Reverse Transcription Kit (Life Technologies, 4368813). Real time polymerase chain reaction (RT-PCR) was carried out on a LightCycler® 480 system (Roche Diagnostics), using the QuantiTect SYBR Green PCR kit (Qiagen, 204143) and oligonucleotide primers from Eurofins MWG Biotech ().
Table 1. List of mouse primer sequences used in quantitative PCR.
Measurement of ROS production by luminol-amplified chemiluminescence
ROS production was measured by the luminol-amplified chemiluminescence method. Briefly, 10Citation6 macrophages from atg5−/− or WT mice were treated with LPS (10 ng/ml), washed and resuspended in 0.5 mL of Hank's balanced salt solution containing 10 μmol/L luminol (Sigma, 123072) and 10 units of horseradish peroxidase (Sigma, P6782). Cells were placed at 37°C in the thermostated chamber of the luminometer (Biolumat LB937; Berthold) and chemiluminescence was recorded during 60 min. ROS production corresponds to the total light emission expressed as cpm counts.
Statistical analysis
Results are expressed as the mean ± standard error of the mean (SEM) and were analyzed by Mann-Whitney test using Prism 5.0 software (GraphPad). p < 0.05 was taken as the minimum level of significance.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Noburu Mizushima for providing Atg5flox/flox mice, Sophia Balustre, Marjorie Collery, and Damien Fois for their help during in vivo experiments and Xavier Decrouy for his expertise in confocal microscopy analyses.
Funding
This work was supported by the INSERM, the Université Paris-Est Créteil, the Université Paris-Diderot, the Investissement d'Avenir program ANR-11-Idex-005–02 Sorbonne Paris Cité Laboratoire d'Excellence Inflammex (SL, JL, JEB), and by grants of the Agence Nationale de la Recherche.
References
- Mallat A, Lotersztajn S. Cellular mechanisms of tissue fibrosis. Five. novel insights into liver fibrosis. Am J Physiol Cell Physiol 2013; 305:C789-99; PMID:23903700; http://dx.doi.org/10.1152/ajpcell.00230.2013
- Ramachandran P, Iredale JP. Liver fibrosis: a bidirectional model of fibrogenesis and resolution. QJM 2012; 105:813-7; PMID:22647759; http://dx.doi.org/10.1093/qjmed/hcs069
- Heymann F, Trautwein C, Tacke F. Monocytes and macrophages as cellular targets in liver fibrosis. Inflamm Allergy Drug Targets 2009; 8:307-18; PMID:19534673; http://dx.doi.org/10.2174/187152809789352230
- Moran-Salvador E, Titos E, Rius B, Gonzalez-Periz A, Garcia-Alonso V, Lopez-Vicario C, Miquel R, Barak Y, Arroyo V, Clària J. Cell-specific PPARgamma deficiency establishes antiinflammatory and anti-fibrogenic properties for this nuclear receptor in nonparenchymal liver cells. J Hepatol 2013; 59:1045-53; PMID:23831119; http://dx.doi.org/10.1016/j.jhep.2013.06.023
- Fallowfield JA, Mizuno M, Kendall TJ, Constandinou CM, Benyon RC, Duffield JS, Iredale JP. Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J Immunol 2007; 178:5288-95; PMID:17404313; http://dx.doi.org/10.4049/jimmunol.178.8.5288
- Mizushima N. Autophagy: process and function. Genes Dev 2007; 21:2861-73; PMID:18006683; http://dx.doi.org/10.1101/gad.1599207
- Czaja MJ. Functions of autophagy in hepatic and pancreatic physiology and disease. Gastroenterology 2011; 140:1895-908; PMID:21530520; http://dx.doi.org/10.1053/j.gastro.2011.04.038
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011; 469:323-35; PMID:21248839; http://dx.doi.org/10.1038/nature09782
- Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008; 456:264-8; PMID:18849965; http://dx.doi.org/10.1038/nature07383
- Castillo EF, Dekonenko A, Arko-Mensah J, Mandell MA, Dupont N, Jiang S, Delgado-Vargas M, Timmins GS, Bhattacharya D, Yang H, et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci U S A 2012; 109:E3168-76; PMID:23093667; http://dx.doi.org/10.1073/pnas.1210500109
- Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, Ting JP, Virgin HW, Kastan MB, Semenkovich CF. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab 2012; 15:534-44; PMID:22440612; http://dx.doi.org/10.1016/j.cmet.2012.02.011
- Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, Robbins J, Martinez J, Tabas I. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab 2012; 15:545-53; PMID:22445600; http://dx.doi.org/10.1016/j.cmet.2012.01.022
- Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 2011; 12:222-30; PMID:21151103; http://dx.doi.org/10.1038/ni.1980
- Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011;469:221-5; PMID:21124315; http://dx.doi.org/10.1038/nature09663
- Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, Sher A, Kehrl JH. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol 2012; 13:255-63; PMID:22286270; http://dx.doi.org/10.1038/ni.2215
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA. et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/10.4161/auto.19496
- Weber LW, Boll M, Stampfl A. Hepatotoxicity and mechanism of action of haloalkanes: carbon tetrachloride as a toxicological model. Crit Rev Toxicol 2003;33:105-136; PMID:12708612; http://dx.doi.org/10.1080/713611034
- Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med 2007; 13:851-6; PMID:17572686; http://dx.doi.org/10.1038/nm1603
- Rider P, Carmi Y, Guttman O, Braiman A, Cohen I, Voronov E, White MR, Dinarello CA, Apte RN. IL-1alpha and IL-1beta recruit different myeloid cells and promote different stages of sterile inflammation. J Immunol 2011; 187:4835-43; PMID:21930960; http://dx.doi.org/10.4049/jimmunol.1102048
- Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, Olefsky JM, Brenner DA, Seki E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 2010; 139:323-34; PMID:20347818; http://dx.doi.org/10.1053/j.gastro.2010.03.052
- Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, Barrieau M, Min SY, Kurt-Jones EA, Szabo G. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest 2012; 122:3476-89; PMID:22945633; http://dx.doi.org/10.1172/JCI60777
- Kamari Y, Shaish A, Vax E, Shemesh S, Kandel-Kfir M, Arbel Y, Olteanu S, Barshack I, Dotan S, Voronov E, et al. Lack of interleukin-1alpha or interleukin-1beta inhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. J Hepatol 2011; 55:1086-94; PMID:21354232; http://dx.doi.org/10.1016/j.jhep.2011.01.048
- Zhan SS, Jiang JX, Wu J, Halsted C, Friedman SL, Zern MA, Torok NJ. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology 2006; 43:435-43; PMID:16496318; http://dx.doi.org/10.1002/hep.21093
- Schuppan D, Kim YO. Evolving therapies for liver fibrosis. J Clin Invest 2013; 123:1887-901; PMID:23635787; http://dx.doi.org/10.1172/JCI66028
- Ding WX, Li M, Chen X, Ni HM, Lin CW, Gao W, Lu B, Stolz DB, Clemens DL, Yin XM. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology 2010; 139:1740-52; PMID:20659474; http://dx.doi.org/10.1053/j.gastro.2010.07.041
- Wang Y, Singh R, Xiang Y, Czaja MJ. Macroautophagy and chaperone-mediated autophagy are required for hepatocyte resistance to oxidant stress. Hepatology 2010; 52:266-77; PMID:20578144; http://dx.doi.org/10.1002/hep.23645
- Wang JH, Ahn IS, Fischer TD, Byeon JI, Dunn WA Jr., Behrns KE, Leeuwenburgh C, Kim JS. Autophagy suppresses age-dependent ischemia and reperfusion injury in livers of mice. Gastroenterology 2011; 141:2188-99; PMID:21854730; http://dx.doi.org/10.1053/j.gastro.2011.08.005
- Ni HM, Bockus A, Boggess N, Jaeschke H, Ding WX. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology 2012; 55:222-32; PMID:21932416; http://dx.doi.org/10.1002/hep.24690
- Bertola A, Park O, Gao B. Chronic plus binge ethanol feeding synergistically induces neutrophil infiltration and liver injury in mice: A critical role for E-selectin. Hepatology 2013; 58:1814-1823; PMID:23532958; http://dx.doi.org/10.1002/hep.26419
- Jaeschke H, Farhood A, Smith CW. Neutrophils contribute to ischemia/reperfusion injury in rat liver in vivo. FASEB J 1990;4: 3355-9; PMID:2253850
- Liu ZX, Han D, Gunawan B, Kaplowitz N. Neutrophil depletion protects against murine acetaminophen hepatotoxicity. Hepatology 2006; 43:1220-30; PMID:16729305; http://dx.doi.org/10.1002/hep.21175
- Bonder CS, Ajuebor MN, Zbytnuik LD, Kubes P, Swain MG. Essential role for neutrophil recruitment to the liver in concanavalin A-induced hepatitis. J Immunol 2004; 172:45-53; PMID:14688308; http://dx.doi.org/10.4049/jimmunol.172.1.45
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature 2009; 458:1131-5; PMID:19339967; http://dx.doi.org/10.1038/nature07976
- Hidvegi T, Ewing M, Hale P, Dippold C, Beckett C, Kemp C, Maurice N, Mukherjee A, Goldbach C, Watkins S, et al. An autophagy enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science 2010; 329:229-32; PMID:20522742; http://dx.doi.org/10.1126/science.1190354
- Hernández-Gea V, Ghiassi-Nejad Z, Rozenfeld R, Gordon R, Fiel MI, Yue Z, Czaja MJ, Friedman SL. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012; 12:938-46; PMID:22240484; http://dx.doi.org/10.1053/j.gastro.2011.12.044
- Mallat A, Lodder J, Teixeira-Clerc F, Moreau R, Codogno P, Lotersztajn S. Autophagy: a multifaceted partner in liver fibrosis. Biomed Res Int 2014; 2014:869390; PMID:25254217
- Stienstra R, Saudale F, Duval C, Keshtkar S, Groener JE, van Rooijen N, Staels B, Kersten S, Müller M. Kupffer cells promote hepatic steatosis via interleukin-1beta-dependent suppression of peroxisome proliferator-activated receptor α activity. Hepatology 2010; 51:511-22; PMID:20054868; http://dx.doi.org/10.1002/hep.23337
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885-9; PMID:16625204; http://dx.doi.org/10.1038/nature04724
- Louvet A, Teixeira-Clerc F, Chobert MN, Deveaux V, Pavoine C, Zimmer A, Pecker F, Mallat A, Lotersztajn S. Cannabinoid CB2 receptors protect against alcoholic liver disease by regulating Kupffer cell polarization in mice. Hepatology 2011; 54(4):1217-26; PMID:21735467; http://dx.doi.org/10.1002/hep.24524
- Teixeira-Clerc F, Julien B, Grenard P, Tran Van Nhieu J, Deveaux V, Li L, Serriere-Lanneau V, Ledent C, Mallat A, Lotersztajn S. CB1 cannabinoid receptor antagonism: a new strategy for the treatment of liver fibrosis. Nat Med 2006; 12:671-6; PMID:16715087; http://dx.doi.org/10.1038/nm1421
- Teixeira-Clerc F, Belot MP, Manin S, Deveaux V, Cadoudal T, Chobert MN, Louvet A, Zimmer A, Tordjmann T, Mallat A, et al. Beneficial paracrine effects of cannabinoid receptor 2 on liver injury and regeneration. Hepatology 2010; 52:1046-59; PMID:20597071; http://dx.doi.org/10.1002/hep.23779