
Abstract
Maintenance of mitochondrial function and energy homeostasis requires both generation of newly synthesized and elimination of dysfunctional mitochondria. Impaired mitochondrial function and excessive mitochondrial content are major characteristics of aging and several human pathophysiological conditions, highlighting the pivotal role of the coordination between mitochondrial biogenesis and mitophagy. However, the cellular and molecular underpinnings of mitochondrial mass homeostasis remain obscure. In our recent study, we demonstrate that DCT-1, the Caenorhabditis elegans homolog of mammalian BNIP3 and BNIP3L/NIX, is a key mediator of mitophagy promoting longevity under stress. DCT-1 acts downstream of the PINK-1-PDR-1/Parkin pathway and is ubiquitinated upon mitophagy-inducing conditions to mediate the removal of damaged mitochondria. Accumulation of damaged mitochondria triggers SKN-1 activation, which initiates a bipartite retrograde signaling pathway stimulating the coordinated induction of both mitochondrial biogenesis and mitophagy genes. Taken together, our results unravel a homeostatic feedback loop that allows cells to adjust their mitochondrial population in response to environmental and intracellular cues. Age-dependent decline of mitophagy both inhibits removal of dysfunctional or superfluous mitochondria and impairs mitochondrial biogenesis resulting in progressive mitochondrial accretion and consequently, deterioration of cell function.
A progressive increase in mitochondrial mass is observed in disparate cell types of diverse organisms during aging and in various human age-related disorders. The molecular basis of this disruption of mitochondrial homeostasis remains elusive. We found that mitochondria gradually accumulate with age in several tissues (neurons, intestine, and muscles) in the nematode C. elegans. Depletion of BEC-1, the nematode homolog of the mammalian general autophagy regulator BECN1, mimics the effect of aging on mitochondrial content in young individuals. Thus, inhibition of autophagy results in mitochondria degradation defects and contributes to the accumulation of mitochondria during aging.
Selective autophagic degradation of mitochondria (mitophagy) mediates the elimination of dysfunctional and/or superfluous mitochondria to maintain energy homeostasis. While mitophagy shares key regulatory factors with the general autophagy pathway, it also involves distinct components, specific for mitochondrial degradation. In mammals, the mitochondrial proteins BNIP3 (BCL2/adenovirus E1B 19kDa interacting protein 3) and BNIP3L/NIX (BCL2/adenovirus E1B 19kDa interacting protein 3-like) function as mitophagy receptors and interact with the autophagosome membrane-associated protein LC3 through a WXXL motif to mediate mitochondrial removal. We identified DCT-1 as the homolog of BNIP3 and BNIP3L/NIX in C elegans. Similar to BEC-1 depletion, deficiency of DCT-1 increases mitochondrial mass. DCT-1 is broadly expressed throughout development; it is an integral membrane protein and localizes on the outer mitochondrial membrane. Moreover, DCT-1 contains a WXXL motif and colocalizes with LGG-1, the nematode homolog of Atg8/LC3, similar to its mammalian counterparts.
To investigate mitophagy and identify conditions that either induce or suppress mitochondrial elimination, we developed 2 composite systems for monitoring mitophagy in vivo. We generated transgenic animals expressing the fluorescent reporter Rosella, which comprises a fast-maturing pH-insensitive DsRed fused to a pH-sensitive GFP variant, targeted to mitochondria (mtRosella). The mtRosella signal is quenched upon delivery of mitochondria to lysosomes during mitophagy. In addition to mtRosella, we generated transgenic animals expressing a mitochondria-targeted GFP together with the autophagosomal membrane protein LGG-1 fused to DsRed, and we monitored animals carrying both mitochondrial and autophagosomal markers, under normal and mitophagy-inducing conditions. We found that mitophagy is stimulated upon exposure to several stressors (low insulin/insulin-like growth factor signaling, heat stress, mitochondrial stress and oxidative stress, among others). Importantly, DCT-1 is required for mitophagy induction.
Mitochondrial depolarization stabilizes PINK1 (PTEN induced putative kinase 1) on the outer mitochondrial membrane, which then recruits the cytosolic E3 ubiquitin ligase PARK2/parkin on the organelle. In turn, PARK2 ubiquitinates several outer mitochondrial membrane proteins, marking damaged mitochondria for degradation. Depletion of PINK-1 or PDR-1, the respective C. elegans PINK1 and PARK2 homologs, diminishes mitophagy upon stress and disrupts mitochondrial physiology. DCT-1, PINK-1 and PDR-1 participate in the same genetic pathway that preserves mitochondrial homeostasis and promotes survival under stress conditions. Indeed, stress resistance conferred by DCT-1 overexpression is abolished in PINK-1- and PDR-1-deficient animals. Further analysis revealed that DCT-1 functions downstream of PINK-1 and PDR-1 to control mitophagy. Thus, mitophagy-deficient animals display pronounced mitochondrial dysfunction, sensitivity to various stressors and abrogation of different life span-prolonging interventions in C. elegans.
Intrigued by these findings, we examined the molecular basis of the genetic interaction between DCT-1, PINK-1, and PDR-1. Notably, we found that DCT-1 ubiquitination is enhanced under oxidative stress, in a PINK-1-dependent manner. Moreover, DCT-1 colocalizes with PDR-1, suggesting that DCT-1 ubiquitination is likely mediated by PDR-1. The role of DCT-1 ubiquitination is not clear. Notably, DCT-1 ubiquitination does not lead to its proteasomal degradation, since DCT-1 protein levels remain unaltered under mitophagy-inducing conditions. In this context ubiquitination could promote DCT-1 function upon mitophagy stimulation. Given that BNIP3 and BNIP3L/NIX function as homodimers in mammals, ubiquitination could induce oligomerization or stabilization of DCT-1 to activate mitophagy under stress conditions. Alternatively, ubiquitination of DCT-1 could cause conformational changes that strengthen its interaction with the autophagosomal protein LGG-1.
Accumulation of damaged mitochondria triggers activation of SKN-1, the nematode NFE2L2/Nrf2 (nuclear factor, erythroid 2-like 2) homolog, in mitophagy-deficient animals. Similar to its mammalian counterpart, SKN-1 is stimulated in response to oxidative stress and drives the expression of several mitochondrial biogenesis genes. Interestingly, the dct-1 gene is among the SKN-1 targets. In addition to SKN-1, DAF-16 also regulates the transcription of dct-1. Consistent with this notion, we found that DCT-1, DAF-16 and SKN-1 function nonredundantly to regulate mitophagy and maintain mitochondrial homeostasis under conditions of oxidative stress. BNIP3 expression is upregulated upon hypoxia, in a HIF1A (hypoxia inducible factor 1, α subunit [basic helix-loop-helix transcription factor])-dependent manner, leading to induction of mitophagy, which confers a prosurvival metabolic adaptation response in mammals. The role of DCT-1 in hypoxia-induced mitophagy remains to be investigated. In addition, it remains to be determined whether DCT-1 is also regulated by HIF-1 under hypoxic conditions in C. elegans.
Elevation of cytoplasmic calcium levels is a consequence of mitochondrial dysfunction in mitophagy-deficient animals. Investigating the role of calcium homeostasis, we found that enhanced SKN-1 transcriptional activity depends on increased cytoplasmic calcium levels upon mitophagy inhibition. In mammals, calcium signaling facilitates mitochondrial biogenesis through a cascade that involves CAMK2 (calcium/calmodulin-dependent protein kinase II). We found that increased cytoplasmic calcium levels modulate SKN-1 activity through the CAMK2 homolog, UNC-43, upon mitochondrial dysfunction. Calcium chelation shortens the life span of mitophagy-deficient animals, further highlighting the critical role of calcium in the coordination of mitophagy and mitochondrial biogenesis.
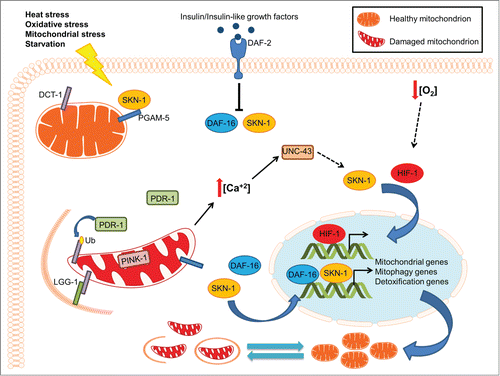
Combined, our findings unravel a key regulatory mechanism that interfaces mitogenesis with the removal of damaged mitochondria through mitophagy. DCT-1 is a nodal element of the pathway integrating environmental and intracellular signals to control mitophagy (). Oxidative stress and the progressive accumulation of damaged mitochondria trigger SKN-1 activation, which in turn initiates a bipartite retrograde signaling pathway, facilitating the coordinated induction of both mitochondrial biogenesis and mitophagy genes. Similar to its mammalian homolog, SKN-1 is tethered to mitochondria through its association with the outer mitochondrial protein PGAM-5. Thus, SKN-1 might function as a central rheostat of mitochondrial homeostasis, sensing mitochondrial damage to promote detoxification and cell survival (). Uncoupling of mitochondrial biogenesis and turnover during aging contributes to the accumulation of dysfunctional mitochondria and consequently, to deterioration of cellular function.
Figure 1. The interplay between mitochondrial biogenesis and mitophagy during aging. Under normal conditions, basal mitophagy flux regulates mitochondrial content depending on the metabolic state of the cell. Under stress conditions, mitophagy induction eliminates damaged mitochondria preserving mitochondrial integrity and promoting cell survival. Excessive mitochondrial damage results in the stimulation of several transcription factors, including SKN-1, DAF-16 and HIF-1, to enhance mitophagy and promote mitochondrial biogenesis by regulating the expression of the DCT-1 mitophagy mediator and several mitochondrial genes. The tight coordination between the opposing processes of mitochondrial biogenesis and removal allows cells to adjust their mitochondrial population in response to extracellular or intracellular signals, preserving mitochondrial function and cell homeostasis.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
Work in the authors' laboratory is funded by grants from the European Research Council (ERC), the European Commission Framework Programmes, and the General Secretariat for Research and Technology of the Greek Ministry of Education (grant ARISTEIA I / Necrosis).