
Abstract
Oncogenic mutation of KRAS (Kirsten rat sarcoma viral oncogene homolog) in colorectal cancer (CRC) confers resistance to both chemotherapy and EGFR (epidermal growth factor receptor)-targeted therapy. We uncovered that KRAS mutant (KRASmut) CRC is uniquely sensitive to treatment with recombinant LGALS9/Galectin-9 (rLGALS9), a recently established regulator of epithelial polarity. Upon treatment of CRC cells, rLGALS9 rapidly internalizes via early- and late-endosomes and accumulates in the lysosomal compartment. Treatment with rLGALS9 is accompanied by induction of frustrated autophagy in KRASmut CRC, but not in CRC with BRAF (B-Raf proto-oncogene, serine/threonine kinase) mutations (BRAFmut). In KRASmut CRC, rLGALS9 acts as a lysosomal inhibitor that inhibits autophagosome-lysosome fusion, leading to autophagosome accumulation, excessive lysosomal swelling and cell death. This antitumor activity of rLGALS9 directly correlates with elevated basal autophagic flux in KRASmut cancer cells. Thus, rLGALS9 has potent antitumor activity toward refractory KRASmut CRC cells that may be exploitable for therapeutic use.
Abbreviations
| ATG5 | = | autophagy-related 5 |
| BAX | = | BCL2-associated X protein |
| BECN1 | = | Beclin 1, autophagy related |
| BRAF | = | B-Raf proto-oncogene, serine/threonine kinase |
| BRAFmut | = | BRAF mutant |
| CASP3 | = | caspase 3, apoptosis-related cysteine peptidase |
| CRC | = | colorectal cancer |
| CRDs | = | carbohydrate recognition domains |
| EGFR | = | epidermal growth factor receptor |
| EPCAM | = | epithelial cell adhesion molecule |
| KRAS | = | Kirsten rat sarcoma viral oncogene homolog |
| KRASmut | = | KRAS mutant |
| LAMP2 | = | lysosomal associated membrane protein 2 |
| LC3B/MAP1LC3B | = | microtubule-associated protein 1 light chain 3 B |
| LGALS9/galectin-9 | = | lectin, galactose-binding, soluble, 9 |
| (serine/threonine kinase) | = | |
| PRKC | = | protein kinase C |
| PS | = | phosphatidylserine |
| PtdIns3K | = | class III phosphatidylinositol 3-kinase |
| RAF1/CRAF | = | Raf-1 proto-oncogene, serine/threonine kinase |
| rLGALS9 | = | recombinant form of LGALS9 |
| RPS6KB1 | = | ribosomal protein S6 kinase, 70kDa, polypeptide 1 |
| SQSTM1/p62 | = | sequestosome 1 |
| TNFSF10/TRAIL | = | tumor necrosis factor (ligand) superfamily, member 10. |
Introduction
Oncogenic mutation of KRAS (Kirsten rat sarcoma viral oncogene homolog) is found in 30–45% of all colorectal cancer (CRC) patients and associates with poor prognosis.Citation1 Specifically, KRAS mutated CRC often poorly responds to chemotherapy due to an increased drug resistance and is insensitive to EGFR (epidermal growth factor receptor)-targeted therapeutics.Citation1,2 Unfortunately, the RAS pathway has proven an elusive target for cancer therapy, with the development of effective drugs targeting RAS being difficult.Citation3 Further, targeting of downstream effectors, such as RAF1, has yielded agents with only modest clinical activity in metastatic CRC patients that is associated with substantial off-target toxicity.Citation4 Therefore, the development of new therapeutic options for this large subset of refractory CRC patients is urgently needed.
Oncogenic KRAS mutation (KRASmut) disrupts apical-basolateral polarity and prevents the formation of tight junctions in colon epithelial cells.Citation5 Loss of cellular polarity is a key factor in the acquisition of metastatic potential and may represent an Achilles' heel of epithelial cancers (reviewed in Citation6,7). Of note, selected members of the galectin family of carbohydrate-binding proteins have recently been recognized as important regulators of epithelial polarity and endocytosis. For instance, LGALS3/Galectin-3 maintains apical polarity by regulating clathrin-independent endocytosis of cargo proteins,Citation8-10 whereas LGALS8/galectin-8 steers endocytic and autophagic removal of bacteria.Citation11
Recently, LGALS9/Galectin-9 (lectin, galactose-binding, soluble, 9) has also emerged as an important regulator of endocytosis and polarity in epithelial cells.Citation12,13 In epithelial monolayers, LGALS9 undergoes a cyclical process of endosomal internalization and rerouting to the apical surface via the trans-Golgi network and recycling endosomes.Citation12,13 Upon targeted deletion of LGALS9 expression in these monolayers, polarity is lost and apical and basolateral proteins mislocalize.Citation12,13 Loss of expression of LGALS9 is also associated with metastatic progression in various epithelial cancers, including breast and colorectal cancer.Citation14-18 Taken together, this suggests that loss of LGALS9 promotes a metastatic tumor phenotype. Conversely, treatment of LGALS9 deficient monolayers with exogenous recombinant LGALS9 (rLGALS9) restores epithelial polarity.Citation12,13 Further, rLGALS9 treatment of murine syngeneic colorectal carcinoma inhibits the formation of lung metastases.Citation19 Thus, treatment with exogenous rLGALS9 could potentially target the postulated Achilles' heel of epithelial cancers.
Here, we show that in colorectal carcinoma cells, exogenous rLGALS9 is rapidly internalized via clathrin- and PRKC (protein kinase C)-, RAF1 (Raf-1 proto-oncogene, serine/threonine kinase)-, MAP2K1 (mitogen-activated protein kinase kinase 1)-dependent endocytosis leading to lysosomal accumulation of rLGALS9. This triggers cell death in refractory KRAS mutant cancer cells, characterized by lysosomal swelling and a halt in the execution of autophagy at the stage of autophagosome-lysosome fusion. Thus, rLGALS9 is a lysosomal inhibitor with potent cytotoxic activity toward refractory KRAS mutant colon carcinoma cells that may be exploitable for therapeutic use.
Results
rLGALS9 internalizes into the lysosomal compartment in nonpolarized cells
LGALS9 maintains apical polarity in established epithelial monolayers through a cyclical process of LGALS9 internalization into early endosomes, routing to the trans-Golgi network, and a resurfacing to the apical cell surface via recycling endosomes.Citation12 In order to follow the routing of LGALS9 in settings of disturbed polarity, nonpolarized MDCK cells, and DLD-1 colorectal cancer cells were treated with rLGALS9/rGAL9(0), a previously reported recombinant form of LGALS9 containing a truncated linker for improved stability.Citation20 Surface binding of fluorescently labeled recombinant rLGALS9 was detected within 1 min, followed by rapid internalization (). Initially, internalized rLGALS9 was localized in close proximity to the cell membrane, but at later time points accumulated in enlarged vesicles more centrally located in the cytoplasm (). This internalization of rLGALS9 was dependent on its carbohydrate recognition domains (CRDs), since the CRD-blocking sugar α-lactose, but not the irrelevant sugar sucrose, abrogated rLGALS9 internalization ().
Figure 1. rLGALS9 is internalized via endosomes and accumulates in the lysosomes. (A) MDCK cells were treated with rLGALS9–594 in the presence or absence of α-lactose (40 mM) or sucrose (40 mM) and confocal images were captured at 1 min, 1 h, and 2 h. (B) Maximum association of rLGALS9–594 with the cell surface of DLD-1 cells at 5 min of incubation (arrows highlight colocalization). Here, DLD-1 cells were incubated with rLGALS9–594, stained with anti-EPCAM-488, fixed with 4% PFA, and subsequently stained with DAPI. (C) For confocal colocalization analysis with early endosomes, DLD-1 cells were transduced with Bacmam CellLight® Fluorescent Protein Construct RAB5A-GFP and incubated with rLGALS9–594 and Hoechst. Maximum colocalization was observed after 15 min (arrows highlight colocalization). (D) For confocal colocalization analysis with late endosomes, DLD-1 cells were transduced with Bacmam CellLight® Fluorescent Protein Construct RAB7A-GFP and incubated with rLGALS9–594 and Hoechst. Maximum colocalization was observed after 45 min (arrows highlight colocalization). (E) For confocal colocalization analysis with the lysosomes, DLD-1 cells were treated with rLGALS9–594, fixed with 4% PFA, and costaining was performed using anti-LAMP2–488. Maximum colocalization was observed after 24 h. (F) Colocalization analysis of EPCAM and rLGALS9 in DLD-1 cells, using Pearson's correlation as determined using ImageJ. Rr = 1, perfect colocalization; Rr = 0, random localization; Rr = −1, total exclusion. (G) Time-dependent increase in lysosomal association of rLGALS9 in DLD-1 cells, determined as the percentage of rLGALS9-positive lysosomes of total amount of lysosomes.
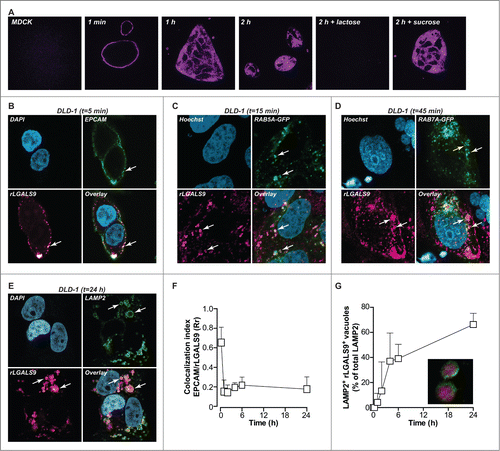
The subcellular localization of rLGALS9 was determined using a panel of cell compartmental markers, which demonstrated that on DLD-1 cells rLGALS9 initially colocalized with the cell surface marker EPCAM (epithelial cell adhesion molecule) (; t = 5 min). In time, this was followed by colocalization of rLGALS9 with the GFP-tagged early endosome marker RAB5A (; t = 30 min), with the GFP-tagged late endosome marker RAB7A (; t = 1 h) and with the lysosomal marker LAMP2 (lysosomal-associated membrane protein 2) ; t = 24 h). Similar intracellular localization of rLGALS9 was observed for MDCK (Fig. S1A-C). Colocalization analysis (using Pearson's correlation coefficient-Rr) confirmed that rLGALS9 very rapidly disappeared from the membrane (), with a time-dependent increase in the percentage of rLGALS9+ LAMP2+ lysosomes ().
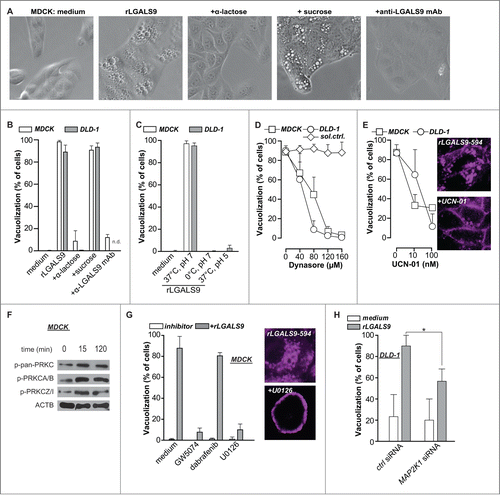
rLGALS9 triggers vacuolization via PRKC-RAF1-MAP2K1-dependent clathrin-mediated internalization
The treatment of MDCK and DLD-1 cells with rLGALS9 was characterized by the progressive formation of large vacuoles (), which affected ∼95% of cells after 24 h (). Vacuole formation was blocked by cotreatment with α-lactose or blocking anti-LGALS9 antibody, but not by sucrose (). Treatment of MDCK or DLD-1 cells on ice, at low pH, or with the DNM/dynamin GTPase inhibitor dynasore abrogated rLGALS9-mediated vacuole formation (, Fig. S2A). Thus, rLGALS9 internalized via active clathrin-dependent endocytosis. To characterize the internalization pathway of rLGALS9, MDCK and DLD-1 cells were co-incubated with inhibitors of kinases involved in endocytosis. Among these, the PRKC-inhibitor UCN-01 dose-dependently reduced rLGALS9 uptake and vacuolization (, Fig. S2A). UCN-01 also reduced colocalization of rLGALS9 with early endosomes (Fig. S2B). In line with this, rLGALS9 treatment phosphorylated various PRKC isoforms, particularly atypical PRKC isoforms PRKCZ/PRKCζ and PRKCI/PRKCλ (). Further, inhibition of RAF1/CRAF (GW5074), but not BRAF (Dabrafenib) strongly reduced rLGALS9-induced vacuolization in MDCK cells (, Fig. S2A). Correspondingly, inhibition of downstream kinases MAP2K1 and MAP2K2 (U0126) also strongly inhibited rLGALS9 internalization and vacuolization (, Fig. S2A). To confirm the requirement for MAP2K-signaling, the predominant MEK isoform in DLD-1, MAP2K1, was knocked down by small interfering (si)RNA (Fig. S2C). Treatment of MAP2K1-deficient DLD-1 cells with rLGALS9 did not induce vacuole formation (, Fig. S2C). Thus, rLGALS9 is internalized via clathrin-dependent, PRKC-RAF1-MAP2K1-mediated endocytosis in nonpolarized MDCK cells and DLD-1 colorectal cancer cells, whereupon it triggers formation of large vacuoles.
Figure 2. rLGALS9 is internalized via clathrin-dependent endocytosis mediated by PRKC and MAP2K1. (A) MDCK cells were treated with rLGALS9 (300 nM) in the presence or absence of α-lactose (40 mM) or sucrose (40 mM) or blocking anti-LGALS9 monoclonal antibody (mAb; 10 μg/ml). Representative light microscopy pictures were taken after 1 h of treatment. (B) Quantification of the percentage of cells having visible vacuoles in MDCK and DLD-1 cells upon 1 h treatment as described in (A). (C) MDCK and DLD-1 cells were treated with rLGALS9 (300 nM) for respectively 1 h and 6 h at 37°C vs. 0°C at pH 7 or pH 5, after which vacuolization was quantified. (D) MDCK and DLD-1 cells were pre-incubated for 1 h with the indicated concentrations of Dynasore (or DMSO solvent control), after which vacuolization induced by rLGALS9 (300 nM) was determined. (E) MDCK and DLD-1 cells were pre-incubated for 1 h with the indicated concentrations of UCN-01 after which vacuolization induced by rLGALS9 (300 nM) was determined. Representative fluorescent pictures (using rLGALS9–594) illustrate the cellular distribution of rLGALS9. (F) Western blot detection of different PRKC isoforms in rLGALS9 (300 nM) treated MDCK cells. (G) MDCK cells were pretreated with RAF1 inhibitor GW5074 (50 μM, 48 h pre-incubation), BRAF inhibitor Dabrafenib (200 nM, 48 h pre-incubation), and MAP2K inhibitor U0126 (100 μM, 2 h pre-incubation) and subsequently treated with rLGALS9 (300 nM), after which the percentage of cells with visible vacuoles was quantified. Representative fluorescence pictures (using rLGALS9–594) illustrate the cellular distribution of rLGALS9 with or without U0126 pretreatment. (H) MAP2K1-deficient DLD-1 cells were treated with rLGALS9 for 6 h after which vacuole formation was quantified.
rLGALS9 has direct dose-dependent cytotoxic activity toward colon carcinoma in vitro and in vivo
Due to the progressive vacuolar swelling upon treatment with rLGALS9, the nuclear size of MDCK and DLD-1 cells was reduced by ∼40% within 5 h (). Treatment of MDCK and DLD-1 cells with rLGALS9 also dose-dependently triggered the apoptotic feature of phosphatidylserine (PS) surface exposure (), loss in cell viability (), reduced sphere formation (), and reduced colony formation (). This cytotoxic effect of rLGALS9 was inhibited by α-lactose, but not sucrose as illustrated for loss of cell viability and PS-exposure (, Fig. S2D). The naturally occurring short and medium LGALS9 isoforms (rLGALS9[S] and rLGALS9[M]) also had cytotoxic effects toward DLD-1 cells, but at higher molar concentrations (, Fig. S2E).
Figure 3. For figure legend, see page 1378.Dose-dependent cytotoxicity of rLGALS9 in vitro and in vivo (A) DAPI staining visualized the nucleus in medium- and rLGALS9-treated (5 h, 300 nM) MDCK cells. (B) Quantification of nuclear size of MDCK and DLD-1 cells after 1- to 5-h treatment with rLGALS9 (300 nM). (C) MDCK and DLD-1 cells were treated with the indicated concentrations of rLGALS9 and analyzed for PS exposure by flow cytometric ANXA5 staining. (D) As in (C), but analyzed for viability after 48 h with the MTS assay. (E) MDCK and DLD-1 cells were plated on nonadherent plates and treated with the indicated concentrations of rLGALS9. The number of tumor spheres was quantified by counting 3 fields-of-view in triplicates after 1 wk. (F) MDCK and DLD-1 cells were plated in Matrigel and treated with the indicated concentrations of rLGALS9. After one wk, colony formation was quantified by counting 3 fields-of-view in triplicates. (G) Analysis of cell viability after 48 h treatment of MDCK and DLD-1 cells with rLGALS9 (300 nM) +/− α-lactose or sucrose (40 mM). (H) Cell viability of DLD-1 cells after 48 h of treatment with increasing doses of rLGALS9/GAL9[0], rLGALS9[S], or rLGALS9[M]. (I) Representative light microscopy pictures of primary patient-derived colorectal cancer cells detailing vacuole formation after 3 h treatment with rLGALS9 (300 nM). (J) Analysis of PS exposure after 24 h treatment of primary patient-derived colorectal cancer cells with rLGALS9 (300 nM) in the presence or absence of α-lactose (40 mM). (K) Analysis of cell viability after 48 h treatment of primary patient-derived colorectal cancer cells with rLGALS9 alone or with rLGALS9 and α-lactose or sucrose (40 mM). (L) Tumor growth of DLD-1 xenografts treated with rLGALS9. In brief, athymic nude mice were subcutaneously injected with 5 × 105 DLD-1 cells in Matrigel. Treatment was started after tumor size reached 100 mm3 and comprised 3 times/wk intraperitoneal injection of rLGALS9 at the indicated doses. After 3 wk, treatment was stopped and survival was determined. Average+/−SEM (M) Kaplan Meyer curve of survival of DLD-1 xenografted mice treated with the indicated concentrations rLGALS9 or solvent control. (N) Weight of rLGALS9-treated mice during the experiment. (O) Representative light microscopy pictures of primary normal colonic epithelial cells with rLGALS9 (300 nM) after 24 h. (P) Analysis of PS exposure after 6 h and cell viability after 48 h of treatment of primary normal colonic epithelial cells with 300 nM rLGALS9.
![Figure 3. For figure legend, see page 1378.Dose-dependent cytotoxicity of rLGALS9 in vitro and in vivo (A) DAPI staining visualized the nucleus in medium- and rLGALS9-treated (5 h, 300 nM) MDCK cells. (B) Quantification of nuclear size of MDCK and DLD-1 cells after 1- to 5-h treatment with rLGALS9 (300 nM). (C) MDCK and DLD-1 cells were treated with the indicated concentrations of rLGALS9 and analyzed for PS exposure by flow cytometric ANXA5 staining. (D) As in (C), but analyzed for viability after 48 h with the MTS assay. (E) MDCK and DLD-1 cells were plated on nonadherent plates and treated with the indicated concentrations of rLGALS9. The number of tumor spheres was quantified by counting 3 fields-of-view in triplicates after 1 wk. (F) MDCK and DLD-1 cells were plated in Matrigel and treated with the indicated concentrations of rLGALS9. After one wk, colony formation was quantified by counting 3 fields-of-view in triplicates. (G) Analysis of cell viability after 48 h treatment of MDCK and DLD-1 cells with rLGALS9 (300 nM) +/− α-lactose or sucrose (40 mM). (H) Cell viability of DLD-1 cells after 48 h of treatment with increasing doses of rLGALS9/GAL9[0], rLGALS9[S], or rLGALS9[M]. (I) Representative light microscopy pictures of primary patient-derived colorectal cancer cells detailing vacuole formation after 3 h treatment with rLGALS9 (300 nM). (J) Analysis of PS exposure after 24 h treatment of primary patient-derived colorectal cancer cells with rLGALS9 (300 nM) in the presence or absence of α-lactose (40 mM). (K) Analysis of cell viability after 48 h treatment of primary patient-derived colorectal cancer cells with rLGALS9 alone or with rLGALS9 and α-lactose or sucrose (40 mM). (L) Tumor growth of DLD-1 xenografts treated with rLGALS9. In brief, athymic nude mice were subcutaneously injected with 5 × 105 DLD-1 cells in Matrigel. Treatment was started after tumor size reached 100 mm3 and comprised 3 times/wk intraperitoneal injection of rLGALS9 at the indicated doses. After 3 wk, treatment was stopped and survival was determined. Average+/−SEM (M) Kaplan Meyer curve of survival of DLD-1 xenografted mice treated with the indicated concentrations rLGALS9 or solvent control. (N) Weight of rLGALS9-treated mice during the experiment. (O) Representative light microscopy pictures of primary normal colonic epithelial cells with rLGALS9 (300 nM) after 24 h. (P) Analysis of PS exposure after 6 h and cell viability after 48 h of treatment of primary normal colonic epithelial cells with 300 nM rLGALS9.](/cms/asset/69ba2d07-add3-44a1-a5f6-fe70988af119/kaup_a_1063767_f0003_c.gif)
In primary patient-derived colon carcinoma cells, treatment with rLGALS9 also induced numerous vacuoles (), resulting in cellular detachment, PS-exposure (, Fig. S2F) and reduced cell viability (), which was inhibited by α-lactose, but not sucrose (). Treatment of mice bearing DLD-1 xenografts with rLGALS9 strongly inhibited tumor growth () and increased overall survival (; 50% for rLGALS9 vs. 0% for control animals), with no detectable signs of toxicity, such as weight loss (). Corresponding to the lack of toxicity in vivo, short-term ex vivo cultures of untransformed primary normal human colon epithelial cells were resistant to rLGALS9, with no vacuolization (), PS-exposure (, Fig. S2F), and no loss in cell viability ().
rLGALS9 selectively induces cell death in KRAS mutant colon carcinoma cells
From the data above it is evident that rLGALS9 triggers prominent cytotoxic effects in DLD-1 colon carcinoma cells characterized by formation of large vacuoles that required RAF1 and MAP2K1 signaling. The cytotoxicity of rLGALS9 similarly depended on MAP2K1 and RAF1, as siRNA-mediated knockdown of either kinase strongly inhibited the cytotoxic effect of rLGALS9 on DLD-1 cells (, Fig. S3A). In contrast, siRNA-mediated knockdown of BRAF did not affect rLGALS9 cytotoxity (, Fig. S3A). Thus, rLGALS9-mediated RAF1, but not BRAF, signaling is required for cytotoxicity. Interestingly, previous studies demonstrated that RAF1 is responsible for transmitting signals from mutated KRAS to downstream kinases MAP2K1 and MAP2K2, whereas BRAF is not required.Citation21 Further, knockdown of RAF1 inhibits tumorigenesis of KRAS mutant cells.Citation22 To investigate whether the cytotoxic activity of rLGALS9 could similarly depend on KRAS mutation, a panel of colon carcinoma cell lines with either oncogenic KRAS or oncogenic BRAF mutation was analyzed. Overnight treatment with rLGALS9 induced PS-exposure and reduced cell viability in all KRASmut BRAFWT cells (), but did not or only minimally affected cell viability of KRASWT BRAFmut cells. Similarly, rLGALS9 reduced colony formation in all KRASmut BRAFWT cell lines, but not in KRASWT BRAFmut cells ().
Figure 4. For figure legend, see page 1380.KRAS mutant but not BRAF mutant colon cancer cell lines are sensitive to rLGALS9 treatment. (A) DLD-1 cells subjected to control siRNA or siRNA-mediated knockdown of MAP2K1, RAF1 or BRAF were treated with 300 nM rLGALS9 for 1 wk, after which cell viability was determined by the MTS assay. (B) A panel of colon carcinoma cell lines with KRAS mutation (KRASmut) or BRAF mutation (BRAFmut) were treated with 300 nM rLGALS9 for 24 h and analyzed for PS exposure. (C) As in (B), but analyzed after 48 h for cell viability with the MTS assay. (D) Colon cancer cell lines were plated in Matrigel and treated with 300 nM rLGALS9. After 72 h, colony formation was quantified by counting 3 fields-of-view in triplicate. (E) KRASWT BRAFWT tumor cell lines Caco-2 and HCT-8 were treated with 300 nM rLGALS9 and analyzed for PS exposure (24 h) and cell viability (48 h). (F-H) The rLGALS9-resistant cell line Caco-2 (KRASWT BRAFWT) was transfected with plasmid encoding KRAS containing the G12V activating mutation. The resulting cell line Caco-2.KRASG12V was analyzed for (F) vacuole formation (G) cell viability and (H) colony formation after treatment with rLGALS9 for 3, 24, 48, and 72 h. (I-J) The rLGALS9-sensitive cell line HCT-8 (KRASWT BRAFWT) was transfected with plasmid encoding KRAS containing the G12V activating mutation or BRAF containing the V600E activating mutation. The resulting cell lines HCT-8.KRASG12V and HCT-8.BRAFV600E were analyzed for (I) vacuole formation and (J) dose-dependent reduction in cell viability. (K) DLD-1 cells were treated with rLGALS9 (300 nM, 24 h) and cell lysates were analyzed by western blot for CASP3 (full-length/cleaved). (L) MDCK and DLD-1 cells were pre-incubated with the pan-caspase inhibitor Z-VAD-fmk (20 μM), the necroptosis inhibitor necrostatin (100 μM) or Z-VAD-fmk+necrostatin and analyzed for rLGALS9-induced vacuolization. (M) Parental HCT116, HCT116.PtdIns3K.WT, HCT116.PtdIns3K.MUT and HCT116.BAX−/− were treated with 300 nM rLGALS9 or 100 ng/ml recombinant human TNFSF10/TRAIL for 24 h, after which cells were analyzed for PS exposure using flow cytometric ANXA5 staining.
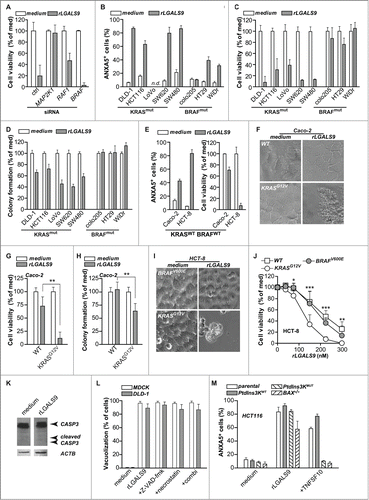
Of note, Caco-2 cells that are both KRAS and BRAF wild-type (KRASWT BRAFWT) proved resistant to rLGALS9, whereas another KRASWT BRAFWT cell line, HCT-8, was sensitive to rLGALS9-mediated cell death (). To identify a causal relationship between expression of a mutant KRAS allele and rLGALS9-mediated cytotoxicity, the rLGALS9 resistant KRASWT BRAFWT cell line Caco-2 was transduced with the KRASG12V oncogene (Caco-2.KRASG12V). In these Caco-2.KRASG12V cells, treatment with rLGALS9 proved cytotoxic, with vacuole formation, reduced cell viability, and inhibited colony formation identical to that induced in other KRASmut BRAFWT cell lines (). Similarly, transduction of the rLGALS9 sensitive KRASWT BRAFWT cell line HCT-8 with the KRASG12V oncogene (HCT-8.KRASG12V) further sensitized these cells to rLGALS9 treatment. In contrast, the oncogenic BRAF mutant allele BRAFV600E (HCT-8.BRAFV600E) did not sensitize cells to rLGALS9 treatment ().
rLGALS9 was previously reported to induce apoptosis in leukemic T-cells, multiple myeloma, Burkitt- and Hodgkin-lymphoma, and melanoma cells.Citation14,23 However, rLGALS9 treatment of DLD-1 cells was not associated with CASP3/caspase-3 processing () nor did the pan-caspase inhibitor Z-VAD-fmk inhibit rLGALS9-induced vacuolization (, Fig. S3B). Furthermore, cell death was not necrotic as cells displayed PS on the surface without membrane permeabilization after 6 h as assessed by propidium iodide (data not shown), nor was cell death inhibited by the necroptosis inhibitor necrostatin alone or in combination with Z-VAD-fmk (, Fig. S3B). To corroborate the apoptosis-independent effect of rLGALS9, isogenic and apoptosis-resistant KRASG12V HCT116 cell lines with an oncogenic class III phosphatidylinositol 3-kinase (PtdIns3K) mutation or with a deficiency in the mitochondrial apoptosis regulator BAX (BCL2-associated X protein) were treated with rLGALS9. Both apoptosis-resistant lines remained sensitive to rLGALS9 cytotoxicity (), but were resistant to apoptosis inducer TNFSF10/TRAIL (tumor necrosis factor [ligand] superfamily, member 10) (). These data indicate that rLGALS9-induced cell death in colon cancer cells with a mutated KRAS allele is likely independent of apoptosis or necrosis.
rLGALS9 induced cell death depends on basal autophagic flux and is associated with lysosomal swelling and autophagosome formation
The progressive accumulation of rLGALS9 within the lysosomal compartment was associated with marked swelling of LAMP2-positive lysosomes in KRASmut cells (, left panels), but not BRAFmut cells (Fig. S4A). This increase in lysosomal size was confirmed using LysoTracker-DND-99 (, right panels), with signal intensity strongly increasing upon rLGALS9 treatment. This swelling of lysosomes was detected in all rLGALS9-sensitive, but not -resistant, cell lines (). Of note, in the wild-type cell lines HCT-8 and Caco-2 the expression of KRASG12V increased the swelling of lysosomes upon rLGALS9 treatment (). Conversely, the expression of BRAFV600E reduced lysosomal swelling in HCT-8 cells (). This rLGALS9-induced swelling of lysosomes correlated with loss in cell viability (). Of note, although the LysoTracker signal as well as the level of red fluorescence of acridine orange-stained cells increased over the first 6 h of treatment with rLGALS9, the signal intensity of both dyes was decreased after 24 h of treatment (Fig. S4B). Analysis of vacuolar diameter revealed that all vacuoles with a diameter >2.5 micrometer in size lost their red acridine orange signal (Fig. S4B), indicating that these dilated lysosomes lost functionality.
Figure 5. rLGALS9-induced cell death is associated with lysosomal swelling and autophagosome formation. (A) Medium control and rLGALS9 (300 nM)-treated DLD-1 cells were stained with anti-LAMP2 (left panel) or the lysosomal marker LysoTracker® Red DND-99. (B) Quantification of the change in LysoTracker signal upon 6 h treatment with 300 nM rLGALS9 of KRASmut cell lines (n = 5), BRAFmut (n = 3), Caco-2WT and Caco-2.KRASG12V, HCT-8WT, HCT-8.KRASG12V and HCT-8.BRAFV600E. rLGALS9-induced lysosomal swelling was quantified by determining fluorescent signal induction using ImageJ (factor = rLGALS9/medium). (C) Linear correlation analysis of the reduction in cell viability upon rLGALS9 treatment and increases in LysoTracker signal upon rLGALS9 treatment. Dotted lines represent the 95% confidence interval. (D) Medium control and rLGALS9 (300 nM) treated DLD-1 cells were stained with anti-LC3B (left panel), monodansylcadaverine (MDC, middle panel), or Cyto-ID (left panel). All these stains detect autophagosomes. (E) Linear correlation analysis of the reduction in cell viability upon rLGALS9 treatment and increases in Cyto-ID signal upon rLGALS9 treatment. Dotted lines represent the 95% confidence interval. (F) A colon cancer cell panel was treated with rLGALS9 (300 nM) for different time points, after which cell lysates were subjected to western blot analysis to detect the lipidated form of LC3B (LC3B-II). (G) Quantification of the fold increase in LC3B-II induced by rLGALS9 in KRASmut cell lines (n = 5), BRAFmut (n = 3), Caco-2WT and Caco-2.KRASG12V (factor = rLGALS9/medium). (H) Western blot analysis of MTOR phosphorylation (serine 2448) and RPS6KB1/p70S6K1 phosphorylation (threonine 389) in KRASmut and BRAFmut cell lines after treatment with 300 nM rLGALS9 for 6 h. (I) DLD-1 cells were subjected to control siRNA or siRNA-mediated knockdown of ATG5 and treated for 1 wk with rLGALS9, after which cell viability was determined using the MTS assay. (J) DLD-1 cells were subjected to control siRNA or siRNA-mediated knockdown of BECN1 and treated for 1 wk with rLGALS9, after which cell viability was determined using the MTS assay. (K) Basal autophagic flux as determined by LC3B-II accumulation upon chloroquine treatment (6 h, 100 µM) in KRASmut cell lines (n = 3), BRAFmut (n = 2), Caco-2 and HCT8. (L) Linear correlation analysis of the increase in LysoTracker signal upon rLGALS9 treatment and basal autophagic flux as determined by chloroquine treatment (6 h, 100 µM) (Fig. S4E). Dotted lines represent the 95% confidence interval. (M) Linear correlation analysis of the decrease in cell viability upon rLGALS9 treatment and basal autophagic flux as determined by 6 h chloroquine treatment. Dotted lines represent the 95% confidence interval.
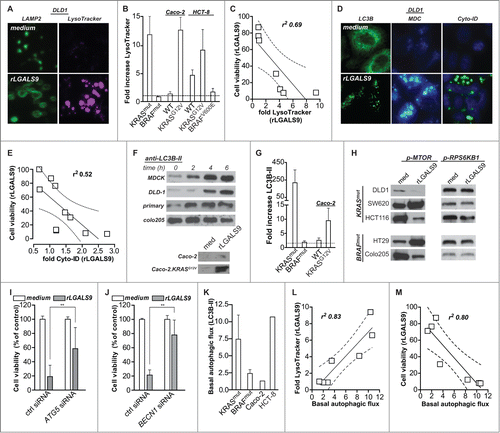
Based on the progressive vacuolization upon rLGALS9 treatment, we reasoned that autophagy might be involved. A hallmark feature of autophagy is the lipidation of MAP1LC3 B/LC3B (microtubule-associated protein 1 light chain 3) into LC3B-II. This LC3B-II is recruited to phagophore membranes yielding a punctate staining pattern. In rLGALS9-treated cells such LC3B puncta were clearly visible (), indicative of autophagosome formation. Correspondingly, the staining intensity of monodansylcadaverine and Cyto-ID, both dyes that can be used to label autophagosomes, were increased in DLD-1 cells upon treatment with rLGALS9 (). This increase in autophagosomes correlated with a loss in cell viability upon rLGALS9 treatment, as illustrated for cyto-ID (). Upon treatment with rLGALS9, the levels of LC3B-II strongly increased in rLGALS9-sensitive KRASmut cell lines, MDCK and primary malignant colorectal cancer cells (, Fig. S4C). In contrast, LC3B-II levels did not increase in rLGALS9-resistant BRAFmut cell lines or KRASWT BRAFwWT Caco-2 cells (), whereas LC3B-II levels did increase in Caco-2.KRASG12V cells (, Fig. S4C). Of note, the formation of autophagosomes during rLGALS9 treatment did not require the autophagy regulator MTOR (mechanistic target of rapamycin [serine/threonine kinase]), as rLGALS9 treatment induced both phosphorylation and dephosphorylation of MTOR in KRASmut and BRAFmut cell lines, with minimal effect on phosphorylation of the downstream MTOR target RPS6KB1 (ribosomal protein S6 kinase, 70kDa, polypeptide 1) ().
To further characterize the role of autophagy in the cytotoxic effects of rLGALS9 in KRASmut colon cancer cells, DLD-1 cells were depleted for ATG5 (autophagy related 5) or the upstream autophagy regulator BECN1/Beclin 1 using siRNA (Fig. S3A), which should block formation of autophagosomes. Indeed, in ATG5-depleted cells LC3B-I accumulated in the absence of conversion into LC3B-II upon rLGALS9 treatment, whereas in control siRNA conditions LC3B-II accumulation was detected (Fig. S4D). Importantly, ATG5 depletion strongly inhibited the cytotoxic activity of rLGALS9 toward DLD-1 cells (). In line with this, siRNA-mediated depletion of BECN1 almost completely abrogated the cytotoxic effect of rLGALS9 treatment in DLD-1 cells (). Thus, inhibition of autophagosome formation inhibits rLGALS9-induced cell death of KRAS mutant colon cancer cells.
Previously, oncogenic mutation of KRAS was shown to associate with increased levels of basal autophagy.Citation24,25 Correspondingly, basal autophagic flux, as determined by the accumulation of LC3B-II upon chloroquine treatment, was high in KRASmut and low in BRAFmut cell lines (, Fig. S4E). In addition, the KRASWT BRAFWT and rLGALS9-sensitive cell line HCT-8 was characterized by high autophagic flux, whereas the KRASWT BRAFWT and rLGALS9-resistant cell line Caco-2 was characterized by low autophagic flux. This basal autophagic flux strongly correlated with increases in LysoTracker signal as well as loss in cell viability upon rLGALS9 treatment (). Thus, the basal autophagic flux in colon cancer cells determines the sensitivity toward rLGALS9 treatment.
rLGALS9 induces dysfunctional autophagy halted at the stage of autophagosomal-lysosomal fusion
During autophagy the accumulation of LC3B-II is typically transient and normalizes when autophagosomes fuse with lysosomes, due to degradation of the autolysosomal content. However, the progressive vacuolization upon rLGALS9 treatment suggested that autophagy may be incompletely executed. To assess this possibility, the levels of LC3B-II and the ubiquitin-binding scaffold protein SQSTM1/p62 (sequestosome 1) were determined over time. Upon full execution of autophagy, e.g. upon rapamycin treatment of DLD-1 cells () and MDCK cells (Fig. S5A), both proteins were degraded after 24 h. However, the levels of LC3B-II and SQSTM1 remained elevated in cells treated with rLGALS9 after 24 h (). These effects of rLGALS9 resembled those of the lysosomal inhibitor chloroquine () and were inhibited by α-lactose but not sucrose (Fig. S5B). Further time course analysis of SQSTM1 levels revealed an initial small decrease in the first hours of rLGALS9 treatment followed by SQSTM1 accumulation at later time points (). This accumulation was observed in all KRASmut BRAFWT cell lines, with an average fold increase in SQSTM1 levels of ∼2.5 (). In contrast, SQSTM1 levels did not change in KRASWT BRAFmut cells ().
Figure 6. Lysosomal accumulation of rLGALS9 results in halted autophagy. (A) LC3B-II levels after 24 h treatment of the indicated cell lines with rLGALS9 (300 nM), autophagy inhibitor chloroquine (100 μM) or autophagy inducer rapamycin (100 nM). (B) Analysis of SQSTM1 levels using ELISA after treatment of DLD-1 as in (A) (factor = rLGALS9/medium). (C) Time-course analysis of SQSTM1 levels in DLD-1 cells treated with rLGALS9 (24 h, 300 nM). (D) SQSTM1 formation induced by rLGALS9 treatment (24 h, 300 nM) in a panel of KRASmut cell lines (n = 5) and BRAFmut (n = 5). (E) Confocal microscopy pictures of DLD-1 cells treated for 24 h with DyLight-594-labeled rLGALS9 and costained with anti-LC3B-488. (F) LC3B/rLGALS9 colocalization was analyzed by Pearson's correlation. (G) Confocal microscopy pictures of DLD-1 cells treated with rLGALS9 (24 h, 300 nM) or chloroquine (24 h, 100 μM) and subsequently stained with anti-LAMP2 and anti-LC3B. LAMP2/LC3B colocalization was analyzed by Pearson's correlation. (H) Confocal microscopy pictures of DLD-1 cells transfected with a mCherry-GFP-LC3 construct treated with rLGALS9 (24 h, 300 nM) or chloroquine (24 h, 100 µM). mCherry/GFP ratio was analyzed using ImageJ software.
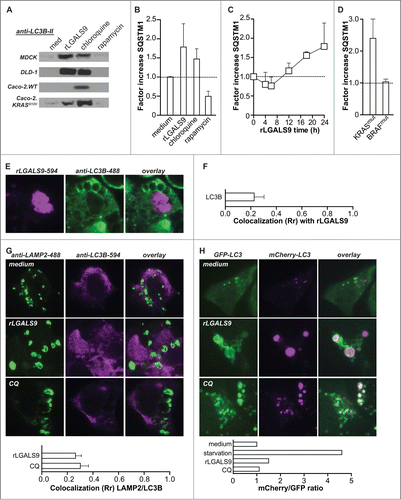
The accumulation of LC3B-II and SQSTM1 suggest that rLGALS9-induced cell death is likely due to inhibition of autophagosome-lysosome fusion. As shown in , rLGALS9 accumulated in lysosomes. In contrast, rLGALS9 overlap with autophagosomes as evaluated by treatment with rLGALS9–594 and counterstaining with LC3B was minimal in DLD-1 cells (), with a corresponding low level of colocalization (). Furthermore, the proteins LAMP2 and LC3B also did not colocalize in rLGALS9-treated cells, indicative of an arrest in the autophagic process at the stage of autophagosome-lysosome fusion (). Of note, treatment with chloroquine triggered an analogous nonoverlapping LAMP2-LC3B staining pattern and Rr colocalization score (). Arrested autophagy upon rLGALS9 treatment was confirmed using mCherry-GFP-LC3-transfected DLD-1 cells in which autophagosome-lysosome fusion will result in loss of GFP signal. Treatment of DLD-1.mCherry-GFP-LC3 with rLGALS9 did not reduce GFP-signal intensity whereby the ratio of mCherry/GFP fluorescent signal did not change, similar to fluorescent LC3-signal after treatment with chloroquine (). Conversely, execution of autophagy in control settings in which DLD-1.mCherry-GFP-LC3 cells were serum starved strongly reduced the GFP-signal intensity and thereby increased the mCherry/GFP ratio (, Fig. S5C).
Taken together, rLGALS9 treatment induces lysosomal swelling and autophagosome formation in KRASmut colon carcinoma cells, which results in halted autophagy by the inhibition of autophagosome-lysosome fusion. Of note, this effect is similar as that induced by the lysosomal inhibitor chloroquine. Thus, rLGALS9 seems to act as an oncogenic KRAS-selective lysosomal inhibitor that induces dysfunctional autophagy and cell death.
Discussion
In this study we show that oncogenic mutation of KRAS sensitizes CRC cells to autophagy-dependent cell death by a recombinant form of the epithelial polarity regulator LGALS9 in vitro and in vivo. We propose a mode of action where rLGALS9 undergoes rapid endosomal internalization in nonpolarized CRC cells and accumulates in the lysosomal compartment. In KRASmut CRC cells, exposure to rLGALS9 is characterized by lysosomal swelling, autophagosome formation and halted autophagosome-lysosome fusion, ultimately leading to cell death (). This sensitivity of KRASmut CRC for rLGALS9 is of potential clinical relevance, since KRASmut CRC cells are notoriously refractory to available therapies, and often acquire therapy resistance by elevating basal levels of autophagy.
Figure 7. rLGALS9 internalization and signaling in CRC. (A) Internalization route of rLGALS9 in polarized epithelial cells. (B) Cell death pathway in nonpolarized KRAS mutant colon carcinoma cells.
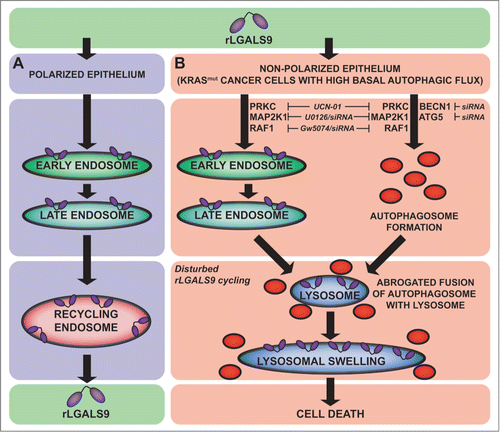
Treatment with rLGALS9 resulted in the accumulation of rLGALS9 in the lysosomal compartment and lysosomal swelling in KRASmut but not BRAFmut CRC cells. Furthermore, cell death induced by rLGALS9 correlated with an increase in levels of autophagosomes. In line with this, rLGALS9-induced cytotoxicity depended on the autophagic pathway, with ATG5 and BECN1 knockdown protecting against rLGALS9 cytotoxicity. However, the execution of autophagy upon treatment of KRAS mut CRC with rLGALS9 was halted at the stage of autophagosome-lysosome fusion, leading to hallmark accumulation of LC3B-II and SQSTM1 over time. Such inhibition of autophagosome-lysosome fusion and LC3B-II-SQSTM1 accumulation has previously been reported for the lysosomal inhibitor chloroquine, leading to vacuolization and lysosomal swelling as identified here for rLGALS9.Citation26,24 Colocalization analysis clearly confirmed lysosomal accumulation of rLGALS9 and lack of association with autophagosomes. Thus, rLGALS9 seems to act as a lysosomal inhibitor, similar to chloroquine, in KRAS mut CRC. Of note, oncogenic KRAS mutation impairs normal lysosomal composition and function in CRC cells, which sensitizes these cells to lysosome-targeting drugs.Citation27
The differential sensitivity of KRASmut and BRAFmut cells to rLGALS9 was directly correlated with their levels of basal autophagic flux, with KRASmut cell lines having high and BRAFmut cells having low basal autophagic flux. In line with this, activated KRAS was previously shown to trigger so-called “autophagy addiction,” with autophagy being required for maintenance of metabolism and tumorigenesis.Citation25,24,28 Conversely, the low autophagic flux levels of BRAFmut cells seem to be specific for CRC, since BRAFmut cells have high basal autophagy levels in other types of cancer such as lung cancerCitation29,30 and melanoma.Citation31 The correlation between high basal autophagic flux levels and the sensitivity toward rLGALS9 treatment may be a logical consequence of the inhibited autophagosome-lysosome fusion that is induced by rLGALS9. By preventing normal execution of autophagy, rLGALS9 induces shortness of energy, which will eventually lead to cell death in such “autophagy-addicted” cancers. In line with this, targeting autophagy has been postulated as a promising target to treat (RAS mutated) cancers with high basal autophagic flux.Citation32 However, cancers which harbor RAS mutations are not always sensitive toward treatment with autophagy inhibitors, and the expression of oncogenic RAS may even promote resistance toward autophagy inhibition in lung cancer cell lines.Citation33 Therefore, the relation between KRASmut expression and sensitivity toward rLGALS9, may be specific for colon cancer.
Besides the maintenance of tumorigenesis, the activation of autophagy is one of the mechanisms by which tumor cells acquire therapy resistance.Citation34 Indeed, high autophagic flux is associated with poor prognosis and drug resistance in CRC,Citation35 with KRAS mut CRC cells being notoriously refractory to available therapies, including EGFR-targeted therapeutics.Citation2 Also in colon cancer stem cells, the autophagic pathway is responsible for the induction of resistance toward photodynamic therapy, which can be counteracted by the use of autophagy inhibitors.Citation36 Similarly, elevated levels of basal autophagic flux are found in cervical cancers compared to healthy tissue, which is even further enhanced by cisplatin treatment.Citation37 Interestingly, simultaneous treatment of ovarian cancers with cisplatin and autophagy inhibitors increases cell death.Citation37 Similar synergistic effects of autophagy inhibitors and standard therapies have been found in many other cancer types.Citation34 Hence, rLGALS9 is not only of clinical relevance in its own right, but may be of particular relevance in combination therapies. Of note, we showed that apoptosis-resistant HCT116 strains with an oncogenic PtdIns3K mutation (found in ∼15–20% of colon cancer patients in large population-based studiesCitation38,39) or BAX-deficiency remained sensitive to rLGALS9 cytotoxicity, while being resistant to the typical apoptosis-inducer TNFSF10/TRAIL. Thus, the autophagy-inhibiting effects of rLGALS9 may be clinically relevant for the treatment of autophagy addicted KRASmut colon cancers and to overcome intrinsic/acquired therapy resistance.
Treatment with rLGALS9 did not negatively affect primary cultures of normal colonic epithelial cells, nor was treatment of xenografted mice with rLGALS9 associated with detectable signs of toxicity. This differential sensitivity of normal versus malignant epithelial cells to rLGALS9 provides a promising therapeutic window. As galectins bind to glycosylated proteins and changes in glycosylation are a hallmark of cancer,Citation40 cancer-specific glycosylation may have altered the receptor profile for rLGALS9. For instance, short and incomplete glycan structures to which rLGALS9 can bind, such as the Forssman antigen,Citation41,12 have been reported in CRC.Citation42,43 Although rLGALS9 did not interact with this glycolipid on DLD-1 cells (data not shown), alternate cancer-specific and de novo glycoepitopes may account for the cancer-selective activity of rLGALS9.
In contrast to the sensitivity of KRASmut CRC cells to rLGALS9, oncogenic mutation of BRAF proved to be associated with resistance to rLGALS9. Correspondingly, siRNA knockdown experiments in KRASmut DLD-1 cells demonstrated that rLGALS9 cytotoxicity did not require BRAF signaling, but was dependent on RAF1 and MAP2K1 signaling. This finding is in line with the reported requirement of oncogenic RAS for RAF1-, but not BRAF-, mediated activation of MAP2K.Citation22,21,44,45 Indeed, although mutated KRAS and BRAF both activate MAP2K/mitogen-activated protein kinase kinases-MAPK/mitogen-activated protein kinase-signaling and disrupt polarity in colon epithelial cells,Citation5 oncogenic KRAS and BRAF transformations typically occur in a mutually exclusive manner.Citation46 Therefore, differences in sensitivity toward rLGALS9 treatment between KRAS and BRAF mutant CRC may rely on their respective signaling via RAF1 and BRAF. Of note, oncogenic KRAS signaling was reported to require heterodimerization between RAF1 and BRAF,Citation47 whereas oncogenic BRAFV600E signaling is activated through monomeric BRAF.Citation48,49 It will be of interest to determine whether a differential requirement for homo- or heterodimerization of RAF1 proteins upon rLGALS9 treatment underlies the differential sensitivity of KRASmut vs. BRAFmut CRC cells.
rLGALS9 is also widely known for its immunomodulatory properties, most notably its inhibition of autoimmune T-cell responses (reviewed in 14). However, rLGALS9 treatment also enhances antitumor immunity in murine sarcoma and melanoma models, leading to prolonged survival.Citation50,51 Further, rLGALS9 can induce activation and expansion of human T-cells ex vivo.Citation52 Conversely, rLGALS9 promotes escape from immune surveillance in Epstein-Barr virus-infected nasopharyngeal carcinoma cells.Citation53 Thus the possible stimulatory or inhibitory effects of rLGALS9 on antitumor immunity in KRASmut CRC will need to be evaluated to fully characterize the therapeutic potential of rLGALS9.
In conclusion, the epithelial polarity regulator rLGALS9 has potent cytotoxic activity toward KRASmut colon cancer by the induction of frustrated autophagy. Therefore, rLGALS9 may be exploitable for therapeutic use in the treatment of refractory KRASmut cancers.
Materials and Methods
Cell lines and primary colorectal (cancer) cells
MDCK, DLD-1, HCT116, HT29, LOVO, SW620, SW480, Caco-2, Colo205, HCT-8, and WiDr were obtained from the American Type Culture Collection (CCL−34, CCL−221, CCL−247, HTB-38, CCL−229, CCL−227, CCL−228, HTB-37, CCL−222, CCL−244, CCL−218). Cell line authenticity was confirmed using STR profiling (Baseclear, The Netherlands and Idexx Radil, USA). All cell lines were cultured in RPMI or DMEM (Lonza, Biowhittaker BE12–604F and BE12–155F) supplemented with 10% fetal calf serum (Hyclone Thermo Scientific, SV30160.03) at 37°C, in a humidified 5% CO2 atmosphere. Primary colorectal cancer cell cultures were obtained by mincing tumor tissue from surgical resection. Primary colonic epithelium was obtained from Tebu-Bio (The Netherlands, 2–96115).
LGALS9 and inhibitors
Recombinant LGALS9 (rLGALS9) containing a truncated 2 amino acid interdomain linker (also known as Gal-9(0)) and the physiologically occurring short isoform of LGALS9, LGALS9(S)/Gal-9(S), were produced as described previously.Citation20 For confocal analysis, rLGALS9 was conjugated to DyLight® 594 following the manufacturer's protocol (DyLight 594 NHS Ester; Piercenet, Thermo scientific, 46412).
The following inhibitors were used: α-lactose (Sigma Aldrich, L3625), sucrose (Merck Millipore, 107651), Dynasore (Sigma Aldrich, D7693), UCN-01 (Sigma Aldrich, U6508), U0126 (Sigma Aldrich, U120), GW5074 (Sigma Aldrich, G6416), necrostatin-1 (Sigma Aldrich, N9037), and Z-VAD-fmk (Calbiochem, 627610). Chloroquine was from the LC3B Antibody Kit for Autophagy (Life Technologies, L10382). LGALS9 blocking antibody was from GalPharma.
Cell transfection and RNA interference
Caco-2.KRASG12V, HCT-8.KRASG12V, and HCT-8.BRAFV600E were generated by transfecting parental Caco-2 and HCT-8 cells with pBabe-Puro-KRAS G12V or pBabe-Puro-BRAF-V600E plasmid (Addgene, plasmid 46746 and plasmid 17544) using Fugene-HD (Promega, E2311). Stable cell lines were obtained by puromycin selection (Gibco/Life Technologies, A11138–03). DLD-1.mCherry-GFP-LC3 was obtained by transfecting DLD-1 cells with GFP-mCherry-LC3 (which was a kind gift of Prof. Andrew Thorburn, Dept. of Pharmacology, University of Colorado Cancer Center) and subsequent puromycin selection.
For RNA interference experiments, cells were precultured in a 12-well plates at a density of 1 × 105 cells/well for 24 h, after which siRNA transfections were performed with Dharmafect-2 (Dharmacon, T-2002–01) following the manufacturer's recommendations using a final concentration of 5 nM siRNA against BRAF (Origine, SR300470), RAF1/CRAF (Origene, SR303972), MAP2K1 (Origene, SR303761), BECN1 (Origene, SR305711) or ATG5 (Origene, SR306286). siRNA-transected cells were maintained under 80% confluence, and used 96 h after transfection for experiments. Cell viability was determined after long-term treatment with rLGALS9 (7 d). Efficiency of knockdown of the target protein was evaluated (96 h after transfection) by western blotting (see below) using anti-BRAF (Santa Cruz Biotechnology, sc-5284), anti- RAF1/CRAF (Sigma Aldrich, R5773), anti-MEK1/2 (Cell Signaling Technology, 9122), anti-BECN1 (Origene, TA301471) and anti-ATG5 (Cell Signaling Technology, 9980), and appropriate secondary HRP-conjugated antibodies (Dako, P0447 and P0448).
Vacuolization experiments
Cells were precultured in a 48-well plate at a density of 2–3 × 104 cells/well. Subsequently cells were treated with rLGALS9 in the presence or absence of pre-incubated inhibitors. Imaging of cell vacuolization (minimal n = 3) of different experimental conditions was determined by digital microscopy (Evos digital inverted microscope), and the degree of vacuolized cells per image was quantified. Percentages of vacuolized cells were calculated based on total cell counts.
Cell viability assays
For ANXA5/annexin-V staining, cells were precultured in a 48-well plate at a density of 2–3 × 104 cells/well. Subsequently cells were treated with rLGALS9 for the indicated concentrations and time span, with or without inhibitors, and subsequently harvested using trypsin. ANXA5 staining was performed according to the manufacturer's protocol (Immunotools, 31490013). In brief, cells were washed once with cold calcium binding buffer and resuspended in calcium buffer containing ANXA5-FITC. After incubation for 10 min at 4°C, PS exposure was analyzed by flow cytometry using a BD Accuri C6 flow cytometer (BD Biosciences) and accessory CFlow Plus analysis software (BD Biosciences).
For long-term viability assays, cells were precultured in a 48-well plate at a density of 1.5 × 104 cells/well. Subsequently, cells were treated with 300 nM rLGALS9 in the presence or absence of 40 mM α-lactose or sucrose, and incubated for 96 h. Then MTS (CellTiter 96® AQueous One Solution Cell Proliferation; Promega, G3580) was added to the culture medium (10% [v/v]) and incubated at 37°C. A maximum death control was induced by treating cells with 20% v/v ethanol. MTS readout was performed by measuring absorbance at 490 nanometer (Versamax microplate reader, Molecular Devices). Absorbance of the maximum death control was subtracted from all values, and cell viability was calculated as percentage of medium control.
Colony-forming and sphere assays
For colony-forming assays, cells were harvested as a single cell suspension using trypsin. BD Matrigel™ (Basement Membrane Matrix; BD bioscience, 354234) was thawed on ice, and RPMI medium supplemented with 10% v/v fetal calf serum was cooled down to 0°C. Per condition, cells were mixed to reach a final solution of 5 × 103 cells in medium containing 3% v/v Matrigel and 300 nM rLGALS9 per well. All conditions were performed in triplicate. Then, cells were placed at 37°C resulting in solidification of the Matrigel, and incubated for 96 h. To quantify colony formation, pictures were taken (minimum of 3 fields of view (FOV) per well, Evos digital inverted microscope), and the number of colonies was counted.
For sphere-forming assays, tumor cells (2 × 103) were cultured in the absence or presence of the indicated concentrations of rLGALS9 in low adherent culture plates (96-wells; Costar, CLS3474). To quantify colony formation, pictures were taken after 1 wk of incubation (minimum 3 FOV per well, Evos digital inverted microscope), and the number of spheres was counted.
Fluorescence microscopy
To determine rLGALS9 uptake and transport, cells were precultured in 8-well borosilicate chamber slides (Lab-Tek II Chambered Coverglass w/Cover 1.5 8 Well; Thermo Scientific, NNU#155409-PK) at a density of 2 × 104 cells/well. Subsequently, cells were transduced with Bacmam CellLight® Fluorescent Protein Constructs RAB5A-GFP or RAB7A-GFP (Life Technologies, C10586 and C10588), and treated with DyLight-coupled rLGALS9 (rLGALS9–594). For other cell compartmental markers, cells were treated with rLGALS9–594 and counterstained with anti-LAMP2 (Abcam, Ab37024) and appropriate secondary antibodies conjugated to Alexa Fluor-488 (Life Technologies, A11034). For cell membrane staining, anti-EPCAM (Stem Cell Technologies, 10109) and secondary antibody conjugated to Alexa Fluor 488 (Life Technologies, A11029) was used. Similar protocols were used in combination with various inhibitors. In the case of intracellular antibody staining, cells were fixed with 4% w/v paraformaldehyde (PFA) and nuclei were stained with DAPI (Sigma Aldrich, D9542). Live-cell imaging was performed on the Solamere Nipkow Confocal Live Cell Imaging system, whereas Z-stacks were obtained using a Leica SP8 Confocal microscope. All fluorescence images and stacks were analyzed using ImageJ software (for colocalization analysis, the Pearson's Correlation plug-in was used) or Leica application suite.
To determine the size of the nucleus, cells were precultured in a 48-well plate at a density of 2–3 × 104 cells/well and treated with rLGALS9. Subsequently, cells were fixed with 4% PFA, and nuclei were stained using DAPI. After 3 washes with PBS, nuclei were evaluated by fluorescence microscopy (Leica) and nuclear size was determined using ImageJ particle analysis plug-ins.
For acridine orange, monodansylcadaverine, LysoTracker staining and cyto-ID, cells were precultured in a 48-well plate at a density of 2–3 × 104 cells/well. After treatment with rLGALS9, acridine orange (1 μg/ml, Sigma Aldrich, A9231), monodansylcadaverine (30 μM, Sigma Aldrich, 30432), LysoTracker® Red DND-99 (1 μM, Life Technologies, L-7528) or cyto-IDhoechst/Hoechst (Cyto-ID autophagy detection kit; Enzo lifesciences, ENZ-51031–0050) was added to the culture medium and subsequently incubated for 30 min at 37°C. Excess dye was removed by 3 repetitive washes using PBS. Each staining was visualized by fluorescence microscopy (Leica) and pictures of each condition were made (minimal n = 3). The corrected total cell fluorescence (CTCF) of LysoTracker signal was determined using ImageJ software. To determine integrity of lysosomes/autophagosomes, cells were stained with acridine orange before rLGALS9 treatment. Of note, pseudocolors were used for all fluorescence microscopic images included in the paper.
Western blot analysis and ELISAs
Cells were precultured in 6-well plates at a density of 3 × 105. Subsequently, cells were treated with rLGALS9 (300 nM) or chloroquine (100 µM) for the indicated time span. Cell lysates were prepared using lysis buffer (150 mM NaCl, 50 mM Tris, pH 7.2, 20 nM EDTA, 20 nM EGTA, 1% NP-40 [Roche, 12879500], 0.1% SDS [Sigma, L6026]; containing Na3VO4 [Sigma, 450243] and protease inhibitor cocktail [Sigmafast; Sigma Aldrich, S8820]), and protein concentration was determined using the Bradford protein assay (Bio-Rad, 500–0006). For each sample, 15–30 µg total protein was loaded onto SDS-PAGE gels (10, 12 or 15%) for electrophoresis. Subsequently proteins were transferred to a nitrocellulose membrane (Amersham Hybond ECL; GE Healthcare, RPN303D). After blocking in 5% (w/v) milk powder/PBS, proteins were detected using primary antibodies against PKCs (Phospho-PKC Antibody Sampler Kit; Cell Signaling Technology, 9921), LC3B (LC3B Antibody Kit for Autophagy, Life Technologies, L10382), CASP3/Caspase-3 (Cell Signaling Technology, 9665), phospho-MTOR (serine 2448; Abcam, Ab109268), or phospho-RPS6KB1/p70S6K1 (threonine 389, Abcam, Ab126818) and appropriate secondary HRP-conjugated antibodies (Dako, P0448). ACTB/β-actin (Directly HRP-conjugated; Abcam, Ab49900) or TUBA/α-tubulin (Acetyl-TUBA, Directly HRP-conjugated, Cell Signaling Technology, (Lys40) (D20G3), 5335) staining were used to assess protein loading. Blots were developed using chemiluminescent substrate (SuperSignal West Dura, Thermo scientific, Life Technologies, 34075), and X-ray films (Super RX, Fujifilm, 47410 19230) or analysis on the ChemiDoc MP system (Bio-Rad). Quantification of detected protein levels was performed using the ImageJ tool for gel analysis.
The lysates used for SQSTM1 ELISAs were prepared as described for western blot analysis. The SQSTM1 ELISAs were performed following the manufacturer's protocol (Enzo Lifesciences, ADI-900–212–0001).
In vivo animal experiment
Experiments involving animals were approved by the Committee for Research and Animal Ethics of the UMCG. Male athymic mice (Hsd:AthymicNude-Foxn1nu, Harlan, The Netherlands) were housed in individual ventilated cages, with ad libitum sterilized water and food, in a 12 h/12 h day and night rhythm. After an acclimatization period of 1 wk, subcutaneous tumors were inoculated by injecting 5 × 105 DLD-1 cells in 100 µl BD Matrigel™. Tumor growth was monitored by measuring tumor volume using a caliper (l·h·w·0.5234). When the tumor reached a mean volume of 0.1 cm3, animals were stratified over different groups (n = 5 per group), and rLGALS9 treatment was started. Treatments with rLGALS9 or PBS control were performed 3 times a wk by intraperitoneal injections. When the tumor reached a size of 1.5 cm3, animals were terminated.
Statistical analysis
Statistical analysis was performed by one-way ANOVA followed by Tukey-Kramer post-test or by 2-sided Student t test using Prism software. P < 0.05 was defined as a statistically significant difference. Where indicated * = P < 0.05; ** = P < 0.01; *** = P < 0.001. All graphs show average +/− standard deviation, unless stated otherwise.
Disclosure of Potential Conflicts of Interest
The authors have read the journal's policy and have the following interests to declare: Drs. Hirashima, Nishi and Niki are board members of GalPharma Co., Ltd. This does not alter the authors' adherence to all Autophagy policies on sharing data and materials.
1063767_supplemental_file.doc
Download MS Word (2.3 MB)Funding
This work was supported by Dutch Cancer Society grants RUG2009–4355 (E.B.), RUG2009–4542, RUG2011–5206, RUG2012–5541, RUG2013–6209 (to E.B./W.H.), the Netherlands Organization for Scientific Research (E.B.), the Melanoma Research Alliance (E.B.), and the Alexander von Humboldt Foundation (E.B/M.B.).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Related Research Data
References
- Phipps AI, Buchanan DD, Makar KW, Win AK, Baron JA, Lindor NM, Potter JD, Newcomb PA. KRAS-mutation status in relation to colorectal cancer survival: the joint impact of correlated tumour markers. Br J Cancer 2013; 108:1757-64; PMID:23511557; http://dx.doi.org/10.1038/bjc.2013.118
- Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012; 486:532-6; PMID:22722830
- Charette N, Vandeputte C, Starkel P. Ras in digestive oncology: from molecular biology to clinical implications. Curr Opin Oncol 2014; 26:454-61; PMID:24849046; http://dx.doi.org/10.1097/CCO.0000000000000088
- Grothey A, Van CE, Sobrero A, Siena S, Falcone A, Ychou M, Humblet Y, Bouche O, Mineur L, Barone C et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013; 381:303-12; PMID:23177514; http://dx.doi.org/10.1016/S0140-6736(12)61900-X
- Magudia K, Lahoz A, Hall A. K-Ras and B-Raf oncogenes inhibit colon epithelial polarity establishment through up-regulation of c-myc. J Cell Biol 2012; 198:185-94; PMID:22826122; http://dx.doi.org/10.1083/jcb.201202108
- Mosesson Y, Mills GB, Yarden Y. Derailed endocytosis: an emerging feature of cancer. Nat Rev Cancer 2008; 8:835-50; PMID:18948996; http://dx.doi.org/10.1038/nrc2521
- Martin-Belmonte F, Perez-Moreno M. Epithelial cell polarity, stem cells and cancer. Nat Rev Cancer 2012; 12:23-38; PMID:NOT_FOUND
- Schneider D, Greb C, Koch A, Straube T, Elli A, Delacour D, Jacob R. Trafficking of galectin-3 through endosomal organelles of polarized and non-polarized cells. Eur J Cell Biol 2010; 89:788-98; PMID:20705359; http://dx.doi.org/10.1016/j.ejcb.2010.07.001
- Straube T, von MT, Honig E, Greb C, Schneider D, Jacob R. pH-dependent recycling of galectin-3 at the apical membrane of epithelial cells. Traffic 2013; 14:1014-27; PMID:23710780; http://dx.doi.org/10.1111/tra.12086
- Lakshminarayan R, Wunder C, Becken U, Howes MT, Benzing C, Arumugam S, Sales S, Ariotti N, Chambon V, Lamaze C et al. Galectin-3 drives glycosphingolipid-dependent biogenesis of clathrin-independent carriers. Nat Cell Biol 2014; 16:595-606; PMID:24837829; http://dx.doi.org/10.1038/ncb2970
- Thurston TLM, Wandel MP, von Muhlinen N, Foeglein A, Randow F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 2012; 482:414-8; PMID:22246324; http://dx.doi.org/10.1038/nature10744
- Mishra R, Grzybek M, Niki T, Hirashima M, Simons K. Galectin-9 trafficking regulates apical-basal polarity in Madin-Darby canine kidney epithelial cells. Proc Natl Acad Sci U S A 2010; 107:17633-8; PMID:20861448; http://dx.doi.org/10.1073/pnas.1012424107
- Mo D, Costa SA, Ihrke G, Youker RT, Pastor-Soler N, Hughey RP, Weisz OA. Sialylation of N-linked glycans mediates apical delivery of endolyn in MDCK cells via a galectin-9-dependent mechanism. Mol Biol Cell 2012; 23:3636-46; PMID:22855528; http://dx.doi.org/10.1091/mbc.E12-04-0329
- Wiersma VR, de BM, Helfrich W, Bremer E. Therapeutic potential of Galectin-9 in human disease. Med Res Rev 2013; 33 Suppl 1:E102-26; PMID:21793015; http://dx.doi.org/10.1002/med.20249
- Irie A, Yamauchi A, Kontani K, Kihara M, Liu D, Shirato Y, Seki M, Nishi N, Nakamura T, Yokomise H et al. Galectin-9 as a prognostic factor with antimetastatic potential in breast cancer. Clin Cancer Res 2005; 11:2962-8; PMID:15837748; http://dx.doi.org/10.1158/1078-0432.CCR-04-0861
- Kageshita T, Kashio Y, Yamauchi A, Seki M, Abedin MJ, Nishi N, Shoji H, Nakamura T, Ono T, Hirashima M. Possible role of galectin-9 in cell aggregation and apoptosis of human melanoma cell lines and its clinical significance. Int J Cancer 2002; 99:809-16; PMID:12115481; http://dx.doi.org/10.1002/ijc.10436
- Liang M, Ueno M, Oomizu S, Arikawa T, Shinonaga R, Zhang S, Yamauchi A, Hirashima M. Galectin-9 expression links to malignant potential of cervical squamous cell carcinoma. J Cancer Res Clin Oncol 2008; 134:899-907; PMID:18264727; http://dx.doi.org/10.1007/s00432-008-0352-z
- Yamauchi A, Kontani K, Kihara M, Nishi N, Yokomise H, Hirashima M. Galectin-9, a novel prognostic factor with antimetastatic potential in breast cancer. Breast J 2006; 12:S196-S200; PMID:16959001; http://dx.doi.org/10.1111/j.1075-122X.2006.00334.x
- Nobumoto A, Nagahara K, Oomizu S, Katoh S, Nishi N, Takeshita K, Niki T, Tominaga A, Yamauchi A, Hirashima M. Galectin-9 suppresses tumor metastasis by blocking adhesion to endothelium and extracellular matrices. Glycobiology 2008; 18:735-44; PMID:18579572; http://dx.doi.org/10.1093/glycob/cwn062
- Nishi N, Itoh A, Fujiyama A, Yoshida N, Araya S, Hirashima M, Shoji H, Nakamura T. Development of highly stable galectins: truncation of the linker peptide confers protease-resistance on tandem-repeat type galectins. FEBS Lett 2005; 579:2058-64; PMID:15811318; http://dx.doi.org/10.1016/j.febslet.2005.02.054
- Blasco RB, Francoz S, Santamaria D, Canamero M, Dubus P, Charron J, Baccarini M, Barbacid M. c-Raf, but not B-Raf, is essential for development of K-Ras oncogene-driven non-small cell lung carcinoma. Cancer Cell 2011; 19:652-63; PMID:21514245; http://dx.doi.org/10.1016/j.ccr.2011.04.002
- Karreth FA, Frese KK, DeNicola GM, Baccarini M, Tuveson DA. C-Raf is required for the initiation of lung cancer by K-Ras(G12D). Cancer Discov 2011; 1:128-36; PMID:22043453; http://dx.doi.org/10.1158/2159-8290.CD-10-0044
- Wiersma VR, de Bruyn M, van Ginkel RJ, Sigar E, Hirashima M, Niki T, Nishi N, Samplonius DF, Helfrich W, Bremer E. The glycan-binding protein galectin-9 has direct apoptotic activity toward melanoma cells. J Invest Dermatol 2012; 132:2302-5; PMID:22572821; http://dx.doi.org/10.1038/jid.2012.133
- Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 2011; 25:460-70; PMID:21317241; http://dx.doi.org/10.1101/gad.2016311
- Kim MJ, Woo SJ, Yoon CH, Lee JS, An S, Choi YH, Hwang SG, Yoon G, Lee SJ. Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J Biol Chem 2011; 286:12924-32; PMID:21300795; http://dx.doi.org/10.1074/jbc.M110.138958
- Yoon YH, Cho KS, Hwang JJ, Lee SJ, Choi JA, Koh JY. Induction of lysosomal dilatation, arrested autophagy, and cell death by chloroquine in cultured ARPE-19 cells. Invest Ophthalmol Vis Sci 2010; 51:6030-7; PMID:20574031; http://dx.doi.org/10.1167/iovs.10-5278
- Fehrenbacher N, Bastholm L, Kirkegaard-Sørensen T, Rafn B, Bøttzauw T, Nielsen C, Weber E, Shirasawa S, Kallunki T, J Jäättelä M. Sensitization to the Lysosomal Cell Death Pathway by Oncogene-Induced Down-regulation of Lysosome-Associated Membrane Proteins 1 and 2. Cancer Res 2008; 68:6623-33; PMID:18701486; http://dx.doi.org/10.1158/0008-5472.CAN-08-0463
- Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, Debnath J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell 2011; 22:165-78; PMID:21119005; http://dx.doi.org/10.1091/mbc.E10-06-0500
- Strohecker AM, White E. Autophagy promotes BrafV600E-driven lung tumorigenesis by preserving mitochondrial metabolism. Autophagy 2014; 10:384-5; PMID:24362353; http://dx.doi.org/10.4161/auto.27320
- Strohecker AM, Guo JY, Karsli-Uzunbas G, Price SM, Chen GJ, Mathew R, McMahon M, White E. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov 2013; 3:1272-85; PMID:23965987; http://dx.doi.org/10.1158/2159-8290.CD-13-0397
- Maddodi N, Huang W, Havighurst T, Kim K, Longley BJ, Setaluri V. Induction of autophagy and inhibition of melanoma growth in vitro and in vivo by hyperactivation of oncogenic BRAF. J Invest Dermatol 2010; 130:1657-67; PMID:20182446; http://dx.doi.org/10.1038/jid.2010.26
- Mancias JD, Kimmelman AC. Targeting autophagy addiction in cancer. Oncotarget 2011; 2:1302-6; PMID:22185891
- Morgan MJ, Gamez G, Menke C, Hernandez A, Thorburn J, Gidan F, Staskiewicz L, Morgan S, Cummings C, Maycotte P et al. Regulation of autophagy and chloroquine sensitivity by oncogenic RAS in vitro is context-dependent. Autophagy 2014; 10:1814-26; PMID:25136801; http://dx.doi.org/10.4161/auto.32135
- Chen S, Rehman SK, Zhang W, Wen A, Yao L, Zhang J. Autophagy is a therapeutic target in anticancer drug resistance. Biochim Biophys Acta 2010; 1806:220-9; PMID:20637264
- Lai K, Killingsworth MC, Lee CS. The significance of autophagy in colorectal cancer pathogenesis and implications for therapy. J Clin Pathol 2014; 67:854-8; PMID:25055793; http://dx.doi.org/10.1136/jclinpath-2014-202529
- Wei MF, Chen MW, Chen KC, Lou PJ, Lin SY, Hung SC, Hsiao M, Yao CJ, Shieh MJ. Autophagy promotes resistance to photodynamic therapy-induced apoptosis selectively in colorectal cancer stem-like cells. Autophagy 2014; 10:1179-92; PMID:24905352; http://dx.doi.org/10.4161/auto.28679
- Leisching G, Loos B, Botha M, Engelbrecht AM. A nontoxic concentration of Cisplatin induces autophagy in cervical cancer: selective cancer cell death with autophagy inhibition as an adjuvant treatment. Int J Gynecol Cancer 2015; 25:380-388; PMID:25695544; http://dx.doi.org/10.1097/IGC.0000000000000365
- Liao X, Morikawa T, Lochhead P, Imamura Y, Kuchiba A, Yamauchi M, Nosho K, Qian ZR, Nishihara R, Meyerhardt JA et al. Prognostic Role of PIK3CA Mutation in Colorectal Cancer: Cohort Study and Literature Review. Clin Cancer Res 2012; 18:2257-68; PMID:22357840; http://dx.doi.org/10.1158/1078-0432.CCR-11-2410
- Barault L, Veyrie N, Jooste V, Lecorre D, Chapusot C, Ferraz JM, Li+¿vre A, Cortet M, Bouvier AM, Rat P et al. Mutations in the RAS-MAPK, PI(3)K (phosphatidylinositol-3-OH kinase) signaling network correlate with poor survival in a population-based series of colon cancers. Int J Cancer 2008; 122:2255-9; PMID:18224685; http://dx.doi.org/10.1002/ijc.23388
- Fuster MM, Esko JD. The sweet and sour of cancer: glycans as novel therapeutic targets. Nat Rev Cancer 2005; 5:526-42; PMID:16069816; http://dx.doi.org/10.1038/nrc1649
- Nagae M, Nishi N, Nakamura-Tsuruta S, Hirabayashi J, Wakatsuki S, Kato R. Structural analysis of the human galectin-9 N-terminal carbohydrate recognition domain reveals unexpected properties that differ from the mouse orthologue. J Mol Biol 2008; 375:119-35; PMID:18005988; http://dx.doi.org/10.1016/j.jmb.2007.09.060
- Ono K, Hattori H, Uemura K, Nakayama J, Ota H, Katsuyama T. Expression of Forssman antigen in human large intestine. J Histochem Cytochem 1994; 42:659-65; PMID:7512587; http://dx.doi.org/10.1177/42.5.7512587
- Hakomori S. Tumor-associated carbohydrate antigens. Annu Rev Immunol 1984; 2:103-26; PMID:6085749; http://dx.doi.org/10.1146/annurev.iy.02.040184.000535
- Preto A, Figueiredo J, Velho S, Ribeiro AS, Soares P, Oliveira C, Seruca R. BRAF provides proliferation and survival signals in MSI colorectal carcinoma cells displaying BRAF(V600E) but not KRAS mutations. J Pathol 2008; 214:320-7; PMID:18098337; http://dx.doi.org/10.1002/path.2295
- Kim JS, Lee C, Foxworth A, Waldman T. B-Raf is dispensable for K-Ras-mediated oncogenesis in human cancer cells. Cancer Res 2004; 64:1932-7; PMID:15026326; http://dx.doi.org/10.1158/0008-5472.CAN-03-3862
- Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature 2002; 418:934-4; PMID:12198537; http://dx.doi.org/10.1038/418934a
- Freeman AK, Ritt DA, Morrison DK. The importance of Raf dimerization in cell signaling. Small GTPases 2013; 4:180-5; PMID:23985533; http://dx.doi.org/10.4161/sgtp.26117
- Freeman AK, Ritt DA, Morrison DK. Effects of Raf Dimerization and Its Inhibition on Normal and Disease-Associated Raf Signaling. Mol Cell 2013; 49:751-8; PMID:23352452; http://dx.doi.org/10.1016/j.molcel.2012.12.018
- Lavoie H, Thevakumaran N, Gavory G, Li JJ, Padeganeh A, Guiral S, Duchaine J, Mao DY, Bouvier M, Sicheri F et al. Inhibitors that stabilize a closed RAF kinase domain conformation induce dimerization. Nat Chem Biol 2013; 9:428-36; PMID:23685672; http://dx.doi.org/10.1038/nchembio.1257
- Nobumoto A, Oomizu S, Arikawa T, Katoh S, Nagahara K, Miyake M, Nishi N, Takeshita K, Niki T, Yamauchi A et al. Galectin-9 expands unique macrophages exhibiting plasmacytoid dendritic cell-like phenotypes that activate NK cells in tumor-bearing mice. Clin Immunol 2009; 130:322-30; PMID:18974023; http://dx.doi.org/10.1016/j.clim.2008.09.014
- Nagahara K, Arikawa T, Oomizu S, Kontani K, Nobumoto A, Tateno H, Watanabe K, Niki T, Katoh S, Miyake M et al. Galectin-9 increases Tim-3+ dendritic cells and CD8+ T cells and enhances antitumor immunity via galectin-9-Tim-3 interactions. J Immunol 2008; 181:7660-9; PMID:19017954; http://dx.doi.org/10.4049/jimmunol.181.11.7660
- Gooden MJ, Wiersma VR, Samplonius DF, Gerssen J, van Ginkel RJ, Nijman HW, Hirashima M, Niki T, Eggleton P, Helfrich W et al. Galectin-9 activates and expands human T-helper 1 cells. PLoS One 2013; 8:e65616-; PMID:23741502; http://dx.doi.org/10.1371/journal.pone.0065616
- Klibi J, Niki T, Riedel A, Pioche-Durieu C, Souquere S, Rubinstein E, Le MS, Guigay J, Hirashima M, Guemira F et al. Blood diffusion and Th1-suppressive effects of galectin-9-containing exosomes released by Epstein-Barr virus-infected nasopharyngeal carcinoma cells. Blood 2009; 113:1957-66; PMID:19005181; http://dx.doi.org/10.1182/blood-2008-02-142596