
Abstract
Autophagy represents an intracellular degradation process which is involved in both regular cell homeostasis and disease settings. In recent years, the molecular machinery governing this process has been elucidated. The ULK1 kinase complex consisting of the serine/threonine protein kinase ULK1 and the adapter proteins ATG13, RB1CC1, and ATG101, is centrally involved in the regulation of autophagy initiation. This complex is in turn regulated by the activity of different nutrient- or energy-sensing kinases, including MTOR, AMPK, and AKT. However, next to phosphorylation processes it has been suggested that ubiquitination of ULK1 positively influences ULK1 function. Here we report that the inhibition of deubiquitinases by the compound WP1130 leads to increased ULK1 ubiquitination, the transfer of ULK1 to aggresomes, and the inhibition of ULK1 activity. Additionally, WP1130 can block the autophagic flux. Thus, treatment with WP1130 might represent an efficient tool to inhibit the autophagy-initiating ULK1 complex and autophagy.
Abbreviations
| ACTB/β-actin | = | actin, β |
| AMBRA1 | = | autophagy/Beclin 1 regulator 1 |
| AMPK | = | AMP-activated protein kinase |
| ATG | = | autophagy related |
| Baf A1 | = | bafilomycin A1 |
| BECN1 | = | Beclin 1, autophagy related |
| Dox | = | doxycycline |
| DUB | = | deubiquitinases |
| EBSS | = | Earle's balanced salt solution |
| GFP | = | green fluorescent protein |
| MAP1LC3/LC3 | = | microtubule-associated protein 1 light chain 3 |
| MBP | = | myelin basic protein |
| MTOR | = | mechanistic target of rapamycin (serine/threonine kinase) |
| MTORC1 | = | MTOR complex 1 |
| PtdIns3K | = | phosphatidylinositol 3-kinase |
| PRKA | = | protein kinase, AMP-activated |
| SQSTM1/p62 | = | sequestosome 1 |
| TRAF6 | = | TNF receptor-associated factor 6, E3 ubiquitin protein ligase |
| TUBE | = | tandem ubiquitin binding entity |
| UCH | = | ubiquitin carboxyl-terminal hydrolase |
| ULK1 | = | unc-51 like autophagy activating kinase 1 |
| USP | = | ubiquitin-specific peptidase |
| WT | = | wild type. |
Introduction
Macroautophagy (hereafter referred to as autophagy) is an intracellular degradation process contributing to the recycling of long-lived, aggregated or misfolded proteins, or even entire organelles. During this process, the cargo to be degraded becomes enveloped within a double-membrane vesicle (referred to as an autophagosome), which is then transported to and fuses with lysosomes. Autophagy occurs at basal levels in most cell lines in order to sustain cellular homeostasis. However, autophagy can also be actively induced upon stress conditions such as nutrient- or energy-depletion, intracellular pathogens, oxidative or ER stress. In the past 2 decades, the molecular understanding of the machinery governing autophagy has substantially increased. Autophagy-related (ATG) gene products mediate all steps of the autophagic flux, including vesicle nucleation, elongation, closure, and fusion with lysosomes.Citation1 Next to ATG proteins, several non-ATG proteins are centrally involved in the regulation of autophagy, including the nutrient- and energy-sensing kinases MTOR (mechanistic target of rapamycin [serine/threonine kinase]) or AMP-activated protein kinase (AMPK).Citation1,2 The ATGs can be grouped into several functional units.Citation1 Two macromolecular protein complexes regulate the initiation of the autophagic process, i.e. the ULK1 protein kinase complex and the class III phosphatidylinositol 3-kinase (PtdIns3K) lipid kinase complex.Citation1 The ULK1 core complex consists of the serine/threonine protein kinase ULK1 (unc-51 like autophagy activating kinase 1) and the adapter proteins ATG13, RB1CC1/FIP200 (RB1-inducible coiled-coil 1), and ATG101. ULK1 is one of the 5 mammalian orthologs of yeast Atg1. In 2009, several groups reported the mechanistic details for how this complex is assembled and receives input from the upstream MTOR complex 1 (MTORC1).Citation3-6 In the current model, MTORC1 associates with the ULK1 complex under nutrient-rich conditions and keeps the complex in an inactive state by phosphorylating ULK1 and ATG13. Upon nutrient depletion, MTORC1 dissociates from this complex and the MTOR-dependent inhibitory ULK1-sites become dephosphorylated. This in turn leads to the activation of ULK1, ULK1 autophosphorylation and ULK1-dependent ATG13-RB1CC1 transphosphorylation.Citation7-11 However, central aspects of this model remain unresolved, including the identification of the MTOR-dependent phospho-sites of ATG13 or the ULK1-dependent phospho-sites of RB1CC1. Additionally, the importance of the ULK1-dependent ATG13 phospho-sites has been challenged by our group.Citation12 Another level of complexity is added to the above described model by the fact that other kinases have been reported to regulate the ULK1 complex, including AMPK and AKT/PKB,Citation13-17 and that alternative post-translational modifications of ULK1 have been described, including acetylation and ubiquitination.Citation18,19 Recently Nazio et al. have shown that AMBRA1, which is a component of the class III PtdIns3K complex, not only becomes phosphorylated by ULK1, but in turn recruits the E3-ligase TRAF6 (TNF receptor-associated factor 6).Citation19 TRAF6 apparently supports Lys63/K63-linked ULK1 ubiquitination, leading to the stabilization and activation of ULK1.Citation19 To date, several ULK1 substrates presumably mediating its proautophagic function have been identified, such as AMBRA1, BECN1, or DAPK3/ZIPK (death-associated protein kinase 3).Citation20-22 Recently, it has been reported that yeast Atg1 phosphorylates Atg9.Citation23 Furthermore, we were able to demonstrate that ULK1 phosphorylates all 3 subunits of AMPK, ultimately leading to the inhibition of AMPK activity.Citation24
Although there are the above-described hints that the ULK1 complex is regulated by ubiquitination, the overall picture as to how this posttranslational modification regulates the ULK1 complex is incomplete. Furthermore, the role of deubiquitinases (DUBs) for the regulation of autophagy is completely uncharacterized. In the present work, we made use of the partially selective DUB inhibitor WP1130 (also known as degrasyn), which has originally been identified during a library screen for small compounds that inhibit IL6-induced phosphorylation of STAT3.Citation25,26 We demonstrate that treatment of cells with this compound leads to the recruitment of ULK1 to aggresomes. This relocalization is most likely caused by an increased ubiquitination status of ULK1. We further observed that ULK1 activity is severely compromised upon ULK1 recruitment to aggresomes. Additionally, treatment with WP1130 leads to the blockade of the autophagic flux. Taken together, we propose that WP1130 represents an efficient tool to inhibit autophagy in general and to modulate ULK1 activity in particular. Next to the direct inhibition of ULK1 kinase activity, the regulation of ULK1 ubiquitination might be an effective approach to modulate the autophagic response.
Results
WP1130 treatment induces the transfer of ULK1 to aggresomes
It has previously been reported that ULK1 function is controlled by ubiquitination.Citation19 Additionally, it has been speculated that the deubiquitinase (DUB) inhibitor WP1130 might increase autophagy.Citation27 Accordingly, we aimed at the in-depth characterization of this compound with regard to autophagy signaling. We made use of the previously described Flp-In™ T-REx™ 293 cells inducibly expressing GFP-ULK1 upon doxycycline treatment.Citation24 Interestingly, incubation with 5 µM WP1130 resulted in decreased levels of GFP-ULK1.() A similar observation was made for GFP-ULK2 (Fig. S1A). Next we investigated whether the reduced GFP-ULK1 protein levels were caused by proteasomal degradation. However, treatment with the proteasome inhibitors MG132 or bortezomib did not abolish the WP1130-induced reduction of GFP-ULK1, indicating that the deubiquitinase inhibitor WP1130 reduces GFP-ULK1 levels without the involvement of the proteasome ()
Figure 1 (See previous page). WP1130 induces transfer of GFP-ULK1 to aggresomes. (A) After induction of GFP-ULK1 expression with doxycycline (Dox) for indicated times, Flp-In™ T-REx™ 293 GFP-ULK1 cells were left untreated (M) or treated with 5 µM WP1130 for 2, 4 or 6 h. Equal protein amounts of cleared cellular lysates were subjected to anti-ULK1 and anti-ACTB/β-actin immunoblotting. (B) After induction of GFP-ULK1 expression with Dox for 3 h, cells were left untreated (M) or treated with 5 µM MG132 or 10 nM bortezomib (Bor), respectively. After 30 min, cells were either lysed directly (0) or 5 µM WP1130 was added for 2, 4, or 6 h. Subsequently cells were lysed and cleared cellular lysates were separated by SDS-PAGE and analyzed by immunoblotting using antibodies against ULK1 and ACTB/β-actin. (C) After induction of GFP-only (upper panels) or GFP-ULK1 (lower panels) expression with Dox for 3 h, Flp-In™ T-REx™ 293 cells were treated for indicated times with 5 µM WP1130 and analyzed by confocal laser scanning microscopy. For GFP-ULK1 expressing cells, zoomed insets are displayed. The GFP-only or GFP-ULK1 signal is displayed in green and the Hoechst signal in blue in the merged images. (D) After induction of GFP-ULK1 or GFP-only expression with Dox for 3 h, Flp-In™ T-REx™ 293 cells were treated with 5 µM WP1130 for the indicated intervals. Following WP1130 treatment, detergent-soluble and -insoluble fractions were prepared and analyzed for ULK1, GFP, and ACTB/β-actin levels by immunoblotting. (A, B and D) Data shown are representative of at least 3 independent experiments. Fold changes were calculated by dividing each normalized ratio (protein to loading control) by the average of the ratios of the control lane (control lane: fold change = 1.00, n ≥ 3). Results are mean ± SD and are given below the corresponding blots.
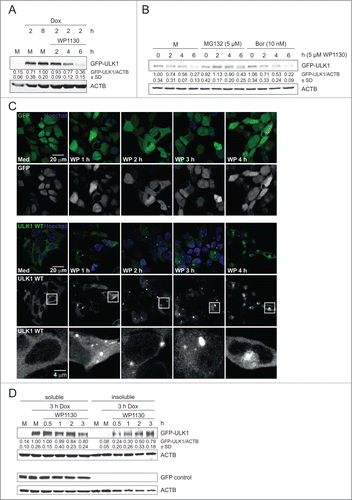
It has been previously reported that WP1130 induces the formation of juxtanuclear aggresomes.Citation27 Additionally, it has been shown that the tyrosine kinases BCR-ABL and JAK2 are recruited to these aggresomes upon WP1130 treatment, which then results in inhibition of their kinase activity.Citation26,28 Accordingly, we speculated that GFP-ULK1 might not be degraded by proteolysis but might be instead transferred to aggresome-like structures upon WP1130 treatment. To test this, we analyzed the effect of WP1130 treatment on the localization of GFP-ULK1 by confocal microscopy. Indeed, incubation with WP1130 resulted in the recruitment of GFP-ULK1 to juxtanuclear punctate structures ( and Fig. S1B). Similarly, GFP-ULK2 was also recruited to these aggresomes upon WP1130 treatment (Fig. S1C). Furthermore, recruitment to aggresomes did not require ULK1 activity since a kinase-dead version of GFP-ULK1 was also translocated to these juxtanuclear punctate structures (Fig. S1C). The WP1130-induced recruitment of GFP-ULK1 to aggresomes was also monitored by live-cell imaging (Movies S1–S3). We observed that small GFP-ULK1 aggregates are already formed upon induction of GFP-ULK1 expression (Movie S1). However, formation of these small peripheral aggregates was clearly enhanced by WP1130 treatment (Movie S2). Furthermore, WP1130 treatment induced the translocation of these aggregates to the perinuclear aggresome (Movie S2). It has been previously reported that microtubule disruption with nocodazole inhibits aggresome formation.Citation29 Indeed, parallel incubation of cells with WP1130 and nocodazole abolished aggresome formation and led to the generation of smaller GFP-ULK1 aggregates in the periphery (Movie S3). Further analysis of the aggresomes revealed that these structures comprised ubiquitin—including K63-linked ubiquitin—and MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3), and to a minor extent contained SQSTM1/p62. (Fig. S2) To confirm these microscopy observations by an independent biochemical assay, we analyzed the WP1130-dependent distribution of GFP-ULK1 in Triton X-100-soluble and -insoluble fractions. Notably, WP1130 treatment led to the redistribution of GFP-ULK1 from the soluble to the insoluble fraction () Taken together, WP1130 treatment does not result in the proteolytic degradation of GFP-ULK1 but induces its transfer to aggresomes.
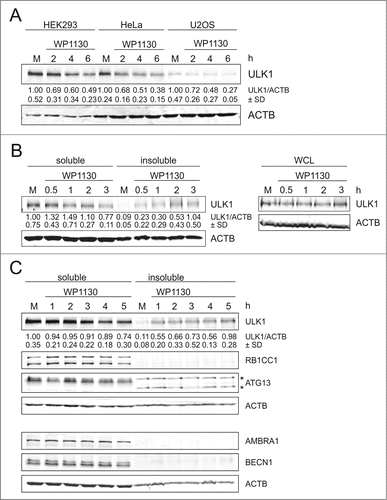
It has been previously observed that overexpression of GFP-ULK1 leads to the formation of cytosolic GFP-positive puncta.Citation30 In order to exclude that the observed effect is just an overexpression artifact, we analyzed the protein levels and distribution of endogenous ULK1 upon WP1130 treatment in 3 different cell lines, i.e., HEK293, HeLa, and U2OS cells (). Again incubation with WP1130 apparently led to strongly reduced ULK1 protein levels in cleared cellular lysates of these cells. This effect could not be reversed by coincubation with different protease inhibitors or with bafilomycin A1, which raises the lysosomal pH by blocking the lysosomal proton pump and thus blocks autophagic degradation (Fig. S3). Additionally and like the GFP-tagged derivative, endogenous ULK1 redistributed from the Triton X-100-soluble to the -insoluble fraction in HEK293 cells (, left panels), whereas overall cellular levels of endogenous ULK1 remained unaltered (, right panels). We next addressed the question whether other components of the ULK1 complex (i.e., ATG13 and RB1CC1/FIP200) or components of the PIK3C3/VPS34-PIK3R4/VPS15-BECN1-ATG14 PtdIns3K complex (i.e., BECN1 and AMBRA1) are redistributed to aggresomes. However, these proteins were not present in the insoluble fraction following WP1130 treatment,() indicating a rather specific process for ULK1. Finally, we investigated whether ULK1 redistribution is a reversible process. For that, we treated HEK293 cells with WP1130 for 3 h, and then resuspended the cells in WP1130-free medium. We observed that the levels of Triton X-100 soluble ULK1 increased again, and that in turn the amount in the insoluble fraction decreased (Fig. S4) This indicates that WP1130-mediated relocalization of ULK1 to aggresomes is indeed a reversible process. Collectively, our results show that WP1130 treatment also results in the recruitment of endogenous ULK1 to aggresomes, but that this is not the case for other autophagy-relevant proteins.
Figure 2. WP1130 induces aggregation of endogenous ULK1. (A) HEK293, HeLa, or U2OS cells were left untreated or were treated with 5 µM WP1130 for 2, 4 or 6 h. Cleared cellular lysates were subjected to anti-ULK1 and anti-ACTB/β-actin immunoblotting. (B) HEK293 cells were left untreated or were treated with 5 µM WP1130 for indicated intervals. Then detergent-soluble or -insoluble fractions, or whole-cell lysates (WCL) were prepared and analyzed by anti-ULK1 and anti-ACTB/β-actin immunoblotting. (C) HEK293 cells were left untreated or were treated with 5 µM WP1130 for indicated intervals. Then detergent-soluble or -insoluble fractions were prepared and analyzed for ULK1, RB1CC1, ATG13, AMBRA1, BECN1 and ACTB/β-actin by immunoblotting. Asterisks indicate unspecific background bands. (A–C) Data shown are representative of at least 3 independent experiments. Fold changes were calculated by dividing each normalized ratio (protein to loading control) by the average of the ratios of the control lane (control lane: fold change = 1.00, n ≥ 3). Results are mean ± SD and are given below the corresponding blots.
WP1130 treatment increases ubiquitination of ULK1
Since WP1130 has been previously identified as deubiquitinase inhibitorCitation26,27 and since the juxtanuclear ULK1-aggresomes obviously contained ubiquitin (Fig. S2), we next asked the question whether the ubiquitination status of ULK1 is affected by this compound. In a first approach, we employed short-term incubation (0.5 h) with WP1130 and analyzed ubiquitination of immunopurified GFP-ULK1 by anti-mono- and poly-ubiquitin immunoblotting () Indeed, GFP-ULK1 ubiquitination increased with WP1130 treatment. In an alternative approach, we transfected Flp-In™ T-REx™ 293 cells expressing either GFP or GFP-ULK1 with cDNA encoding HA-ubiquitin and analyzed immunopurified GFP and GFP-ULK1 by anti-HA immunoblotting. Whereas we did not detect ubiquitinated proteins from GFP-expressing cells, we again observed increased GFP-ULK1 ubiquitination following WP1130 treatment () In order to characterize the type of ubiquitin linkage, we transfected HEK293 cells with cDNAs encoding different HA-ubiquitin variants and incubated the cells with WP1130. Analysis of GFP-Trap®-purified GFP-ULK1 by anti-HA immunoblotting indicated that ubiquitination of ULK1 is mainly established by non-K48-, non-K63-linked ubiquitin chains () In order to confirm ULK1 ubiquitination by an alternative approach, we purified ubiquitinated proteins from cellular lysates using tandem ubiquitin binding entity (TUBE) technology. Treatment of HEK293 cells or Flp-In™ T-REx™ 293 cells with WP1130 increased the amount of purified ULK1 or GFP-ULK1 compared to untreated cells ().
Figure 3. DUB inhibition leads to increased GFP-ULK1 ubiquitination. (A) After induction of GFP-ULK1 expression with Dox for 3 h, Flp-In™ T-REx™ 293 cells were incubated in full medium (M) with or without 5 µM WP1130 for 0.5 h. Subsequently, cells were lysed and GFP-immunopurification was performed. Purified GFP-ULK1 was analyzed by anti-ULK1 and anti-ubiquitin (P4D1) immunoblotting. ULK1 signal is red and ubiquitin signal is green in the merged image. (B) Flp-In™ T-REx™ 293 cells were transiently transfected with cDNA encoding HA-ubiquitin 24 h prior to WP1130 treatment. After induction of GFP or GFP-ULK1 expression with Dox for 3 h, cells were incubated in full medium (M) with or without 5 µM WP1130 for 1.5 h. Subsequently, cells were lysed and GFP-immunopurification was performed. Purified GFP (left panels) and GFP-ULK1 (right panels) were analyzed by anti-GFP, anti-ULK1 and anti-HA immunoblotting. GFP or GFP-ULK1 signal is red and HA signal is green in the merged images. (C) Flp-In™ T-REx™ 293 cells were transiently transfected with cDNAs encoding different HA-ubiquitin variants (WT, KallR, K48only, K63only, K48/63only, K48R, K63R, K48/63R) 24 h prior to WP1130 treatment. After induction of GFP-ULK1 expression with Dox for 3 h, cells were incubated in full medium (M) with or without 5 µM WP1130 for 1.5 h. Subsequently, cells were lysed and GFP-immunopurification was performed. Purified GFP-ULK1 was analyzed by anti-ULK1 and anti-HA immunoblotting. ULK1 signal is red and HA signal is green in the merged image. (D) HEK293 cells or GFP-ULK1-expressing Flp-In™ T-REx™ 293 cells were left untreated or were treated with 5 µM WP1130 for indicated intervals. Subsequently, cells were lysed and immunopurification of ubiquitinated proteins was performed using agarose-TUBE2. Purified ubiquitinated proteins were analyzed by anti-ULK1 (as indicated) or anti-ubiquitin (FK2) immunoblotting.
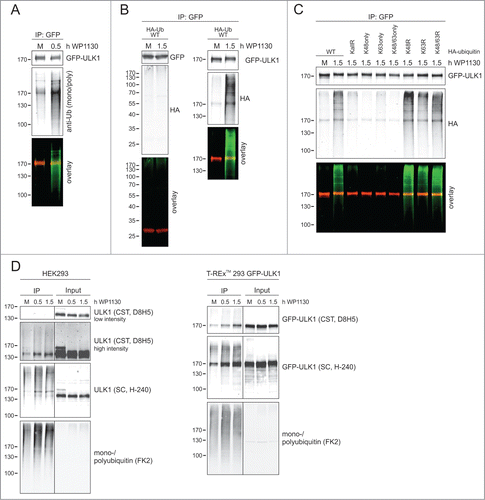
It has been previously reported that WP1130 targets the deubiquitinases (DUBs) USP5, USP9X, USP14, UCHL1 and UCHL5.Citation27 In order to test whether these DUBs are involved in the regulation of ULK1 expression levels, we transfected HEK293 cells simultaneously with the corresponding siRNAs and analyzed endogenous ULK1 expression after 48 and 72 h, respectively (Fig. S5A) However, we could not detect any alterations of ULK1 expression levels. Since USP9X is one major target of WP1130,Citation27,28,31-33 we also analyzed the effect of WP1130 on ULK1 expression levels in USP9X−/o HCT116 cells.Citation34 WP1130 reduced ULK1 levels in both USP9X+/o and USP9X−/o HCT116 cells to a similar extent (Fig. S5B) indicating that USP9X is not the major target of WP1130 with regard to ULK1 regulation. In order to identify the DUB(s) targeted by WP1130, we performed a DUB profiling assay with WP1130 using a library of 35 recombinant DUBs (DUBProfiler™ Single Point Screening, Ubiquigent, Dundee, UK). However, none of the tested DUBs was significantly inhibited by 1 or 10 µM WP1130 (Fig. S6) Notably, the 35 DUBs also contained the 5 enzymes previously reported to be inhibited by WP1130. Finally, we wanted to confirm that the effect caused by WP1130 is due to the inhibition of DUBs. For that, we employed the pan-DUB inhibitor PR619. This inhibitor transferred ULK1 from the soluble to the insoluble fraction similar to WP1130 (Fig. S7A and B) In contrast, the more selective DUB inhibitors LDN 57444 (targeting UCHL1) and Spautin-1 (targeting USP10 and USP13) did not result in a similar reduction of ULK1 in the soluble fraction (Fig. S7A) Importantly, all analyzed DUB inhibitors did not induce caspase activity in the relevant time frame (Fig. S8) In summary, we showed that WP1130 increases ubiquitination of GFP-ULK1. Although this effect is likely caused by the inhibition of DUBs, the previously identified WP1130 targets are apparently not involved.
WP1130-induced recruitment to aggresomes reduces ULK1 kinase activity
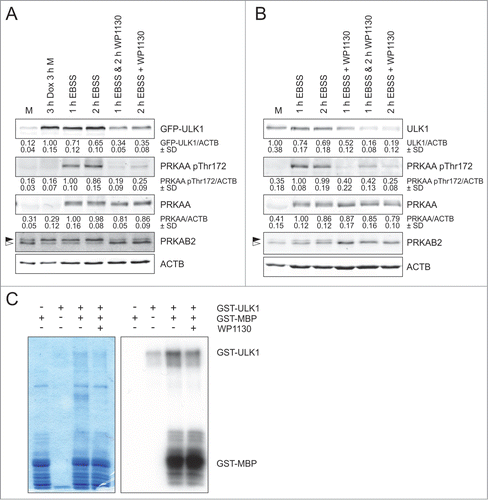
Since we observed that WP1130 treatment leads to the transfer of ULK1 to aggresomes, we next analyzed whether this process also affects ULK1 kinase activity. We have previously identified PRKAB2/AMPK-β2 as direct ULK1 substrate.Citation24 Thus, we analyzed the phosphorylation status of PRKAB2/AMPK-β2 by immunoblotting. For GFP-ULK1, induction of ectopic over-expression is sufficient to increase ULK1 kinase activity () For endogenous ULK1, we starved HEK293 cells in EBSS in order to induce ULK1 kinase activity () WP1130 treatment reduced PRKAB2/AMPK-β2 phosphorylation as detected by a faster migration in SDS-PAGE () We also analyzed phosphorylation of PRKAA/AMPK-α at Thr172. Our group has previously shown that phosphorylation of this activating site is indirectly regulated by ULK1.Citation24 WP1130 treatment clearly reduced PRKAA/AMPK-α phosphorylation at Thr172 in GFP-ULK1-expressing () or wild-type HEK293 cells () without affecting total PRKAA/AMPK-α levels. Taken together, these observations suggest that the WP1130-mediated transfer of ULK1 to aggresomes negatively regulates ULK1 activity. In order to exclude the possibility that WP1130 functions as a direct ULK1 kinase inhibitor, we performed an in vitro kinase assay with recombinant GST-ULK1 and the substrate GST-myelin basic protein (MBP) in the absence and presence of WP1130. WP1130 did not influence ULK1-dependent phosphorylation of MBP () This observation is supported by a kinase screen with WP1130, which was performed by the MRC Protein Phosphorylation Unit in Dundee (UK). None of the 141 tested kinases (including ULK1 and ULK2) were significantly inhibited by WP1130. The data can be found online at http://www.kinase-screen.mrc.ac.uk/screening-compounds/591459. Collectively, these results demonstrate that WP1130 negatively regulates ULK1 activity indirectly through ULK1 aggregation.
Figure 4. ULK1 activity is inhibited by WP1130. (A) After induction of GFP-ULK1 expression with Dox for 3 h, Flp-In™ T-REx™ 293 cells were incubated in full medium (M) or starvation medium (EBSS) for 1 or 2 h in the presence or absence of 5 µM WP1130 for the indicated intervals. Subsequently, cells were lysed and cleared cellular lysates were separated by SDS-PAGE and analyzed by immunoblotting using antibodies against ULK1, PRKAA/AMPK-α (pThr172), PRKAA/AMPK-α, PRKAB1/2 (AMPK-β1/2) and ACTB/β-actin. (B) HEK293 cells were treated and analyzed as in (A). (C) For an in vitro kinase assay, 1 µg GST-myelin basic protein (MBP) was incubated with 0.5 µg GST-ULK1 in the absence or presence of 5 µM WP1130. As controls, GST-MPB or GST-ULK1 were incubated alone. After Coomassie staining of the gels (left panel), autoradiography was performed (right panel). (A and B) Solid arrowheads indicate the phosphorylated form of PRKAB2/AMPK-β2; open arrowheads indicate the unphosphorylated form. Data shown are representative of at least 3 independent experiments. Fold changes were calculated by dividing each normalized ratio (protein to loading control) by the average of the ratios of the control lane (control lane: fold change = 1.00, n ≥ 3). Results are mean ± SD and are given below the corresponding blots.
WP1130 blocks the autophagic flux
It has been previously speculated that WP1130 might increase autophagy.Citation27 Since we observed reduced ULK1 activity upon WP1130 treatment, we next wanted to determine the effect of this compound on the autophagic flux. For that, we applied 3 independent readouts. First, we analyzed localization of endogenous LC3 in HEK293 cells by immunofluorescence. Starvation of the cells in EBSS resulted in an increase of LC3 puncta, which was further increased by treatment with bafilomycin A1 () This effect was completely blocked by WP1130. All WP1130-treated cells showed similar LC3 puncta numbers, and there was no further increase with EBSS, indicating that autophagy is blocked at an early stage of the pathway. Second, we analyzed LC3 turnover in HEK293 cells by immunoblotting (). This assay essentially confirmed the results obtained by LC3 immunofluorescence. All lysates derived from cells incubated with WP1130 showed similar LC3-II levels, and again there was no further increase with EBSS treatment. Of note, WP1130 treatment alone increased LC3-II levels. Third, we performed a long-lived protein degradation assay in HEK293, HeLa and U2OS cells () In all 3 cell lines, WP1130 significantly reduced the degradation of long-lived proteins. As a control we employed 3-methyladenine (3-MA), which is an inhibitor of the PtdIns3K complex and accordingly also blocks an initial step of the autophagic machinery. In summary, WP1130 efficiently blocks the autophagic flux in HEK293, HeLa, and U2OS cells.
Figure 5 (See previous page). The autophagic flux is inhibited by WP1130. (A) HEK293 cells were treated with full medium (Med) or starvation medium (EBSS) in the presence or absence of 20 nM bafilomycin A1 (Baf A1) or 5 µM WP1130 for 2 h and were visualized by confocal laser scanning microscopy. The LC3 signal is displayed in red and the Hoechst signal in blue in the merged images. At least 168 cells were scored for each condition. The number of LC3 puncta and the number of cells per image were quantified using CellProfiler analysis software. Data represent mean ± SD; **P < 0.01 (Student t test, 2-sample assuming unequal variances). (B) HEK293 cells were incubated in full medium (Med) or starvation medium (EBSS) in the absence or presence of 20 nM bafilomycin A1 (Baf A1) and/or 5 µM WP1130 for 2 h. Equal protein amounts from cleared cellular lysates were analyzed for LC3 and GAPDH by immunoblotting. Data shown are representative of at least 3 independent experiments. Fold changes were calculated by dividing each normalized ratio (protein to loading control) by the average of the ratios of the control lane (control lane: fold change = 1.00, n ≥ 3). Results are mean ± SD and are depicted in a bar diagram. **P < 0.01 (Student t test, 2-sample assuming unequal variances) (C) Cellular proteins of HEK293, HeLa and U2OS cells were labeled with L-[14C]valine as described in the Materials and Methods section. Cells were washed and treated with indicated medium (control or EBSS) and inhibitors (5 µM WP1130, 100 nM bafilomycin A1, 10 mM 3-methyladenine) for 4 h. For each sample, the radioactivity of the acid-soluble fraction of the medium and the radioactivity in the cells remaining in the well were measured. Percent degradation was assessed as the acid-soluble radioactivity of the medium divided by the total radioactivity. Data shown are mean of triplicates ± SD; **P < 0.01 (Student t test, 2-sample assuming unequal variances).
![Figure 5 (See previous page). The autophagic flux is inhibited by WP1130. (A) HEK293 cells were treated with full medium (Med) or starvation medium (EBSS) in the presence or absence of 20 nM bafilomycin A1 (Baf A1) or 5 µM WP1130 for 2 h and were visualized by confocal laser scanning microscopy. The LC3 signal is displayed in red and the Hoechst signal in blue in the merged images. At least 168 cells were scored for each condition. The number of LC3 puncta and the number of cells per image were quantified using CellProfiler analysis software. Data represent mean ± SD; **P < 0.01 (Student t test, 2-sample assuming unequal variances). (B) HEK293 cells were incubated in full medium (Med) or starvation medium (EBSS) in the absence or presence of 20 nM bafilomycin A1 (Baf A1) and/or 5 µM WP1130 for 2 h. Equal protein amounts from cleared cellular lysates were analyzed for LC3 and GAPDH by immunoblotting. Data shown are representative of at least 3 independent experiments. Fold changes were calculated by dividing each normalized ratio (protein to loading control) by the average of the ratios of the control lane (control lane: fold change = 1.00, n ≥ 3). Results are mean ± SD and are depicted in a bar diagram. **P < 0.01 (Student t test, 2-sample assuming unequal variances) (C) Cellular proteins of HEK293, HeLa and U2OS cells were labeled with L-[14C]valine as described in the Materials and Methods section. Cells were washed and treated with indicated medium (control or EBSS) and inhibitors (5 µM WP1130, 100 nM bafilomycin A1, 10 mM 3-methyladenine) for 4 h. For each sample, the radioactivity of the acid-soluble fraction of the medium and the radioactivity in the cells remaining in the well were measured. Percent degradation was assessed as the acid-soluble radioactivity of the medium divided by the total radioactivity. Data shown are mean of triplicates ± SD; **P < 0.01 (Student t test, 2-sample assuming unequal variances).](/cms/asset/68a2432a-e9ed-436a-8467-e7373c97fe87/kaup_a_1067359_f0005_c.gif)
Taken together, we showed that WP1130 targets ULK1/2 for ubiquitination and subsequent transfer to aggresomes, and thereby reduces the overall ULK1/2 enzymatic activity. Furthermore, the WP1130-mediated functional knockdown of ULK1/2 is accompanied with a reduced autophagic potential.
Discussion
The mammalian ULK1-ATG13-RB1CC1-ATG101 complex is centrally involved in the initiating steps of autophagy. In recent years, the molecular regulation of this complex has been deciphered. Generally, major attention has been attributed to the phosphorylation of the components of this complex. However, additional posttranslational modifications have been suggested to play an important role for ULK1 complex regulation, including acetylation and ubiquitination.Citation18,19 Here we report that the treatment of cells with the DUB inhibitor WP1130 leads to increased ULK1 ubiquitination and subsequent recruitment of ULK1 to aggresome-like structures. This ultimately results in reduced ULK1 activity. Furthermore, WP1130 treatment blocks the autophagic flux in HEK293, HeLa, and U2OS cells. Collectively, we hypothesize that the regulation of ULK1 ubiquitination might represent an efficient strategy to modulate autophagy in general and the ULK1 complex in particular.
The observed WP1130 effect might be caused by the fact that the proteasome cannot handle the increased abundance of ubiquitinated proteins in general and of ubiquitinated ULK1 in particular. Accordingly, ULK1 is transported to aggresomes due to proteasomal overload. An alternative possibility is that ULK1 is indeed regulated by a specific ubiquitin conjugation-deconjugation cycle, which becomes disrupted by WP1130-treatment. However, these 2 possibilities are not mutually exclusive and cannot be easily distinguished. Generally, non-K63 ubiquitin chains accumulate upon proteasome inhibition.Citation35 In turn, it has been previously reported that WP1130 treatment leads to the accumulation of JAK2-containing K63-linked ubiquitin chains or generally to the accumulation of cellular proteins containing both K48- and K63-linked polyubiquitin chains.Citation26,27 We observe that ULK1 is recruited to aggresome-like structures containing K63-linked ubiquitin. In addition, we found that immunopurified GFP-ULK1 apparently contains non-K48-, non-K63-linked ubiquitin chains. However, it has to be kept in mind that the “ubiquitin barcode” of ULK1 might vary during the trafficking from soluble fractions (which was analyzed in our experiments) to insoluble structures.
Recently, Nazio et al. have reported that MTOR controls ULK1 ubiquitination and function through the class III PtdIns3K complex component AMBRA1 and the associated E3-ligase TRAF6.Citation19 In their model, MTOR inhibits AMBRA1 under normal growth conditions through phosphorylation. Upon autophagy induction, AMBRA1 becomes dephosphorylated and supports TRAF6-mediated ULK1 ubiquitination. The authors show K63-linked ULK1 ubiquitination, but they do not comment on the lysine residues being ubiquitinated. Generally, Nazio et al. report ULK1 stabilization and self-association upon ubiquitination. Whereas the latter observation might be in line with ours, we observe a prominent decline in ULK1 activity upon ubiquitination, which is in contrast to their results. Like Nazio et al., we were not able to identify the ubiquitinated lysine residues of ULK1 so far. In addition to TRAF6, it has been reported that the RFWD2/COP1 E3 ligase (ring finger and WD repeat domain 2) interacts with the ULK1 complex.Citation36 Presumably this interaction is mediated via RB1CC1. However, the authors do not observe any alterations of ULK1 expression upon ectopic expression of RFWD2.Citation36 It is likely that additional E3 ligases targeting ULK1 will be identified in the future.
The human genome encodes 79 DUBs which are predicted to be catalytically active, and they can be subdivided into 5 subfamilies.Citation37 Apparently, the reported WP1130 targets are not, or at least not only, involved in the regulation of ULK1. Especially USP9X has been established as target of WP1130.Citation27,28,31-33 However, ULK1 levels in the soluble fraction remained unaltered in cells which are deficient for USP9X or in which USP9X has been knocked down by siRNA. Furthermore, LC3 turnover occurred normally in USP9X-negative HCT116 cells, but was blocked by WP1130 treatment in both wild-type (WT) and USP9X-negative cells (data not shown). Taken together, it is well conceivable that additional DUBs or a combination of DUBs mediate the WP1130 effect on ULK1. Of note, the pan-DUB inhibitor PR619 phenocopied the effect of WP1130, indicating that ULK1 accumulation is indeed caused by the inhibition of DUBs. Accordingly, future studies need to reveal the exact positions of the post-translationally modified lysine residues of ULK1, the enzymes involved in ubiquitin conjugation/deconjugation, and the subtype of ubiquitin linkage in order to establish a more complete model of ubiquitin-dependent ULK1 regulation.
It is a difficult approach to determine whether the inhibitory effect of WP1130 on autophagy is solely mediated through the alteration of ULK1/2 ubiquitination, aggregation, and activity. The usage of genetic ULK1/2 knockout systems does not represent an entirely feasible approach since autophagy is largely blocked in these cell models. So far we can only say that putative ULK1/2-independent autophagy pathways are not equally affected by WP1130 (data not shown). Furthermore, of the analyzed proteins so far (i.e., ULK1, ATG13, RB1CC1, BECN1, and AMBRA1), only ULK1 is redistributed to aggresomes upon WP1130 treatment. Nevertheless, future studies will have to show whether additional components of the autophagic machinery are regulated by DUBs and thus are potentially involved in the WP1130-mediated inhibition of autophagy.
The ULK1 complex is tightly controlled by a network of upstream nutrient- or energy-sensing kinases, including AKT, MTOR, and AMPK. All these kinases directly phosphorylate ULK1 and thus act to regulate the ULK1 complex (reviewed in ref. 2, 7-11). Accordingly, different small compounds targeting these kinases modulate autophagy, e.g., rapamycin (inhibition of MTOR) or resveratrol (activation of AMPK). Notably, these substances generally induce autophagy. With regard to the inhibition of the autophagic flux, current compounds either target later steps of the autophagic machinery (e.g., bafilomycin A1, chloroquine), or the PIK3C3/VPS34-BECN1 complex (e.g., 3-methyladenine). As a serine/threonine kinase, ULK1 represents an appropriate target for pharmacological modulation, and recently the structure of the ULK1 kinase domain in complex with multiple inhibitors has been reported.Citation38 We propose that the development of more specific inhibitors of ULK1-deubiquitinating enzymes and thus the modulation of ULK1 ubiquitination might represent an alternative strategy to regulate ULK1 function, next to direct inhibition of the kinase activity. Generally, it has been proposed that MTOR inhibitors and other anticancer drugs induce cytoprotective autophagy, ultimately leading to a compromised efficacy of these compounds.Citation11 Therefore, combinatorial therapies employing these agents and parallel ULK1 inhibition might represent a promising anticancer strategy.
Materials and Methods
Cells
HEK293 cells, HeLa cells, U2OS cells, and Flp-In™ T-REx™ 293 cells (R780-07, Invitrogen, Life Technologies) inducibly expressing GFP-ULK1 WT, GFP-ULK1 kinase-dead (kd), GFP-ULK2 WT or GFP-only were cultured in DMEM (4.5 g/l D-glucose, L-glutamine [Gibco, Life Technologies, 41965-039]), supplemented with 10% fetal calf serum, 50 U/ml penicillin and 50 µg/ml streptomycin (full medium). Expression of the respective fusion protein was induced with 0.1 µg/ml doxycycline (Takara, Clontech, 631311). Wild-type and USP9X−/o HCT116 cells (previously described in ref. 34 and kindly provided by Fred Bunz, The Kimmel Cancer Center) were cultured in McCoy 5A medium (PromoCell, C-73220) supplemented with 10% fetal calf serum, 50 U/ml penicillin and 50 µg/ml streptomycin. For amino acid starvation, HEK293 cells and derivatives were washed once and incubated in EBSS (Gibco, Life Technologies, 14155-048) supplemented with 1.8 mM CaCl2 and 0.813 mM MgSO4 for the indicated times.
Antibodies, recombinant proteins, and reagents
Anti-ACTB/β-actin (clone AC-74, A5316), anti-ATG101 (SAB4200175), anti-ATG13 (SAB4200100), and anti-TUBA4A/α-tubulin (clone B-5-1-2, T5168) were purchased from Sigma-Aldrich; anti-BECN1 (11427) and anti-ULK1 (H-240, sc-33182) from Santa Cruz Biotechnology; anti-RB1CC1/FIP200 (A301-574A) from Bethyl Laboratories; anti-AMBRA1 (pab0224-P) from CovalAb S.A.S.; anti-GAPDH (clone 6C5, ab8245), anti-HA (clone 12CA5, ab16918), and anti-LMNB1/lamin B1 (ab16048) from Abcam; anti-PRKAA/AMPKα (2793), anti-phospho-PRKAA/AMPK-α Thr172 (2535), anti-PRKAB1/2 (AMPK-β1/2) (4150), anti-LC3B (2775), anti-ULK1 (clone D8H5, 8054), and anti-ubiquitin (clone P4D1, 3936) from Cell Signaling Technology; anti-ubiquitin (clone FK2, 04-263) and anti-ubiquitin K63-specific (clone Apu3, 05-1308) from Merck Millipore; anti-LC3 (PM036) and anti-SQSTM1/p62 (PM045) from MBL International; and anti-GFP (11814460001) from Roche Diagnostics. IRDye®680- and IRDye®800-conjugated secondary antibodies (926-68020/21 and 926-32210/11) were provided by LI-COR Biosciences. Alexa Fluor® 568-coupled donkey anti-rabbit IgG (H+L) (A10042) or donkey anti-mouse IgG (H+L) (A10037) and Alexa Fluor® 594-coupled goat anti-rabbit IgG (H+L) (A11037) were purchased from Molecular Probes, Life Technologies. Hoechst 33342 (H1399) was purchased from Life Technologies; bafilomycin A1 (B1793), NEM (E3876), pepstatin A (77170), and 3-methyladenine from Sigma-Aldrich; puromycin (ant-pr-1) from InvivoGen (San Diego, CA, USA); Bortezomib (5.043.140.001), calpeptin (03-34-0051), cathepsin Inhibitor III (219419), MG132 (474790), Q-VD-OPh (551476), and Spautin-1 (567569) from Calbiochem, Merck Millipore; and PR619 (SI9619) and LDN 57444 (SI9639) from LifeSensors. WP1130 was purchased from Calbiochem, Merck Millipore (681685) or Axon Medchem BV (1779). GFP-Trap® coupled to agarose beads (gta-200) was purchased from ChromoTek. Agarose-TUBE2 was purchased from LifeSensors (UM402). Staurosporine (BML-EI156) was obtained from Enzo Life Sciences. [14C]Valine (NEC291EU050UC) was purchased from PerkinElmer. GST-ULK1 (SRP5205) and GST-MPB (SRP5096) were purchased from Sigma-Aldrich. [32P]Adenosine 5’-triphosphate (SRP-301) was provided by Hartmann Analytic.
Expression constructs and transfections
Cloning of the cDNAs encoding human ULK1 or the kinase-dead mutant (D165A) of ULK1 into pcDNA5/FRT/TO-GFP and the generation of the corresponding Flp-In™ T-REx™ 293 cells has been previously described.Citation24 Human ULK2 cDNA was amplified from Jurkat J16 cells and cloned into pcDNA5/FRT/TO-GFP. This vector was cotransfected with pOG44 into Flp-In™ TREx™ 293 cells (Invitrogen, Life Technologies, R780-07). Stable transfectants were selected with 200 μg/ml hygromycin B (Invitrogen, Life Technologies, 10687-010) and 5 μg/ml blasticidin (Invitrogen, Life Technologies, A11139-02). pCDNA3.1-based vectors encoding different HA-Ubiquitin variants (WT, KallR, K48R, K63R, K48/63R, K48only, K63only, and K48/63only) were obtained from https://mrcppureagents.dundee.ac.uk (generated by the MRC Protein Phosphorylation and Ubiquitylation Unit, University of Dundee, UK). For transient transfection, typically 10-cm diameter dishes of GFP-ULK1 Flp-In™ T-REx™ 293 cells were cultured, and were transfected with 14 µg DNA using FuGENE® HD transfection reagent (Roche Diagnostics, 04709713001)
Immunoblotting and immunopurification
HEK293 cells or derivatives were incubated in full medium or EBSS supplemented with reagents and time points as indicated. Whole-cell lysates were prepared by heating cell pellets in 2x Laemmli reducing sample buffer for 5 min at 95°C. Detergent-soluble and detergent-insoluble fractions were obtained by lysing cells in lysis buffer (50 mM Tris-HCl, pH 7.5, 1 mM EDTA, 1% [v/v] Triton X-100 [Carl Roth GmbH + Co. KG, 3051.2], 1 mM Na3VO4, 50 mM NaF, 0.1% [v/v] DTT, 150 mM NaCl, Protease Inhibitor Cocktail [Sigma-Aldrich, P2714]) for 30 min on ice and centrifuged for 10 min at 20,000 g at 4°C. The supernatant fraction was used as the detergent-soluble fraction and the residual pellet fraction was heated in 2x Laemmli buffer for 5 min at 95°C and used as the detergent-insoluble fraction. In the case of the detergent-soluble fraction, equal protein amounts (as determined by Bradford assay) were separated on SDS-PAGE followed by standard immunoblot analysis. For the preparation of whole-cell lysates and detergent-insoluble fractions the cell number was adjusted. To immunopurify GFP-ULK1, cells were lysed in the above-mentioned lysis buffer for detergent-soluble fractions (additionally containing 2 mM NEM) for 30 min on ice and centrifuged for 10 min at 20,000 g at 4°C. Immunopurification was carried out for 16 h at 4°C with rotation after addition of GFP-Trap® beads. The agarose beads were washed 3 times with lysis buffer containing 2 mM NEM, heated for 5 min at 95°C in 2× Laemmli buffer and used for immunoblotting. Affinity purification of ubiquitinated proteins was carried out with Agarose-TUBE2 as described in the user's manual. The lysis buffer was supplemented with 2 mM NEM.
In vitro kinase assay
For in vitro phosphorylation, 1 μg GST-MBP was incubated with 0.5 µg GST-ULK1 in 50 mM Tris-HCl, pH 7.5, 0.1 mM EGTA, 0.1 mM DTT, 5 mM Mg(CH3COO)2 and 0.1 mM [32P]ATP in the absence or presence of 5 µM WP1130. The reaction was stopped by the addition of SDS sample buffer after 30 min at 30°C, and was then subjected to SDS-PAGE. After Coomassie staining of the gel, autoradiography was performed.
Confocal laser scanning microscopy and LC3 puncta quantification
HEK293 cells or Flp-In™ T-REx™ 293 cells inducibly expressing GFP-ULK1 WT, GFP-ULK1 kd, GFP-ULK2 WT, or GFP-only were seeded on poly-L-lysine coated cover slips. Expression of the respective fusion protein was induced with 0.1 µg/ml doxycycline for 3 or 16 h. Incubation of cells was performed in media and for times as indicated. Cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS; Gibco, Life Technologies, 14190-094) buffer for 15 min. For analysis of GFP fusion proteins, cells were directly embedded in Mowiol 4-88 (Carl Roth GmbH + Co. KG, 0713.1) containing 1 µg/ml Hoechst. For immunofluorescent staining of endogenous LC3, ubiquitin, K63-linked ubiquitin and SQSTM1/p62, HEK293 cells were washed 3 times with PBS, incubated in 50 mM NH4Cl for 10 min and incubated in PBS for another 5 min. Cells were permeabilized with 0.05% saponin (Sigma-Aldrich, 47036-50G-F) in PBS for 5 min and incubated for 1 h with the corresponding primary antibodies diluted in 0.05% saponin in PBS. Cells were washed 3 times with 0.05% saponin in PBS and incubated for 1 h with the appropriate secondary antibodies. After washing 2 times with 0.05% saponin in PBS and once with PBS, coverslips were embedded in Mowiol 4-88 containing 1 µg/ml Hoechst. For superresolution microscopy cover slips were embedded in VECTASHIELD mounting medium with DAPI (Vector Laboratories, H-1200). Cells were analyzed on a Zeiss LSM 710, Zeiss LSM 780 or Zeiss Elyra PS microscope (Oberkochen, Germany). Hoechst, DAPI, GFP, Alexa Fluor® 568 and Alexa Fluor® 594 were excited at 405 nm, 488 nm, 561 nm and 594 nm, respectively. The number of LC3 puncta and number of cells per image were quantified using CellProfiler analysis software.
Long-lived protein degradation assay
Cells were incubated for 72 h with 0.125 µCi/ml L-[14C]valine-supplemented medium, followed by 2 washes and a 16-h chase in fresh medium containing 10 mM nonradioactive L-valine to allow degradation of short-lived proteins. Next, the cells were washed and treated with the indicated medium and inhibitors for 4 h. For each sample, the radioactivity of the acid-soluble fraction of the medium and the radioactivity in the cells remaining in the well were measured.
Statistical analysis
For western blotting, fold changes were calculated by dividing each normalized density ratio (protein of interest to loading control) by the average of the density ratios of the control lane (control lane: fold change = 1.00, n ≥ 3). Results are mean ±SD and are given below the corresponding blots or are depicted in a bar diagram. For LC3 immunofluorescence, at least 168 cells were scored for each condition. The number of LC3 puncta and the number of cells per image were quantified using CellProfiler analysis software. Data represent mean ± SD and are depicted in a bar diagram. For the long-lived protein degradation assay, the radioactivity of the acid-soluble fraction of the medium and the radioactivity in the cells remaining in the well were measured for each sample. Percent degradation was assessed as the acid-soluble radioactivity of the medium divided by the total radioactivity. Data shown are mean of triplicates ± SD and are depicted in a bar diagram. For all analyses, P values were determined by the Student t test (2-samples, unequal variances) and the significance levels were set as follows: * indicates P < 0.05, ** indicates P < 0.01, *** indicates P < 0.001.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Fred Bunz for providing USP9X+/o and USP9X−/o HCT116 cells, and the MRC Protein Phosphorylation and Ubiquitylation Unit (University of Dundee, UK) for providing anti-USP9X antibodies and cDNAs encoding HA-ubiquitin variants. We thank Stefanie Weidtkamp-Peters and the Center for Advanced Imaging (CAi) of the Heinrich-Heine University Düsseldorf for their support with confocal laser scanning microscopy. We would like to thank Trond Lamark, Terje Johansen, Ian Ganley, and Philip Cohen for helpful discussions.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
1067359_supplemental_movies__3_.zip
Download Zip (7.9 MB)Funding
This work was supported by the Deutsche Forschungsgemeinschaft STO 864/3-1 and STO 864/4-1 (to B.S.), the Research Committee of the Medical Faculty of the Heinrich-Heine-University Düsseldorf 58/2013 (to B.S.), and the Düsseldorf School of Oncology (to S.W. and B.S.; funded by the Comprehensive Cancer Center Düsseldorf/Deutsche Krebshilfe and the Medical Faculty of the Heinrich-Heine-University Düsseldorf).
Related Research Data
References
- Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 2011; 27:107-32; PMID:21801009; http://dx.doi.org/10.1146/annurev-cellbio-092910-154005
- Alers S, Löffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 2012; 32:2-11; PMID:22025673; http://dx.doi.org/10.1128/MCB.06159-11
- Chan EY, Longatti A, McKnight NC, Tooze SA. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol Cell Biol 2009; 29:157-71; PMID:18936157; http://dx.doi.org/10.1128/MCB.01082-08
- Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 2009; 284:12297-305; PMID:19258318
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 2009; 20:1981-91; PMID:19211835; http://dx.doi.org/10.1091/mbc.E08-12-1248
- Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009; 20:1992-2003; PMID:19225151; http://dx.doi.org/10.1091/mbc.E08-12-1249
- Alers S, Löffler AS, Wesselborg S, Stork B. The incredible ULKs. Cell Commun Signal 2012; 10:7; PMID:22413737; http://dx.doi.org/10.1186/1478-811X-10-7
- Chan EY. mTORC1 phosphorylates the ULK1-mAtg13-FIP200 autophagy regulatory complex. Sci Signal 2009; 2:pe51; PMID:19690328; http://dx.doi.org/10.1126/scisignal.284pe51
- Chan EY, Tooze SA. Evolution of Atg1 function and regulation. Autophagy 2009; 5:758-65; PMID:19411825; http://dx.doi.org/10.4161/auto.8709
- Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol 2010; 22:132-9; PMID:20056399; http://dx.doi.org/10.1016/j.ceb.2009.12.004
- Wong PM, Puente C, Ganley IG, Jiang X. The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy 2013; 9:124-37; PMID:23295650; http://dx.doi.org/10.4161/auto.23323
- Alers S, Löffler AS, Paasch F, Dieterle AM, Keppeler H, Lauber K, Campbell DG, Fehrenbacher B, Schaller M, Wesselborg S, et al. Atg13 and FIP200 act independently of Ulk1 and Ulk2 in autophagy induction. Autophagy 2011; 7:1423-33; PMID:22024743; http://dx.doi.org/10.4161/auto.7.12.18027
- Bach M, Larance M, James DE, Ramm G. The serine/threonine kinase ULK1 is a target of multiple phosphorylation events. Biochem J 2011; 440:283-91; PMID:21819378; http://dx.doi.org/10.1042/BJ20101894
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Sci 2011; 331:456-61; PMID:21205641; http://dx.doi.org/10.1126/science.1196371
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132-41; PMID:21258367; http://dx.doi.org/10.1038/ncb2152
- Mack HI, Zheng B, Asara JM, Thomas SM. AMPK-dependent phosphorylation of ULK1 regulates ATG9 localization. Autophagy 2012; 8:1197-214; PMID:22932492; http://dx.doi.org/10.4161/auto.20586
- Shang L, Chen S, Du F, Li S, Zhao L, Wang X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci U S A 2011; 108:4788-93; PMID:21383122; http://dx.doi.org/10.1073/pnas.1100844108
- Lin SY, Li TY, Liu Q, Zhang C, Li X, Chen Y, Zhang SM, Lian G, Ruan K, Wang Z, et al. GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Sci 2012; 336:477-81; PMID:22539723; http://dx.doi.org/10.1126/science.1217032
- Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, Gretzmeier C, Dengjel J, Piacentini M, Fimia GM, et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol 2013; 15:406-16; PMID:23524951; http://dx.doi.org/10.1038/ncb2708
- Di Bartolomeo S, Corazzari M, Nazio F, Oliverio S, Lisi G, Antonioli M, Pagliarini V, Matteoni S, Fuoco C, Giunta L, et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J Cell Biol 2010; 191:155-68; PMID:20921139; http://dx.doi.org/10.1083/jcb.201002100
- Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 2013; 15:741-50; PMID:23685627; http://dx.doi.org/10.1038/ncb2757
- Tang HW, Wang YB, Wang SL, Wu MH, Lin SY, Chen GC. Atg1-mediated myosin II activation regulates autophagosome formation during starvation-induced autophagy. EMBO J 2011; 30:636-51; PMID:21169990 ; http://dx.doi.org/10.1038/emboj.2010.338
- Papinski D, Schuschnig M, Reiter W, Wilhelm L, Barnes CA, Maiolica A, Hansmann I, Pfaffenwimmer T, Kijanska M, Stoffel I, et al. Early steps in autophagy depend on direct phosphorylation of atg9 by the atg1 kinase. Mole Cell 2014; 53:471-83; PMID:24440502; http://dx.doi.org/10.1016/j.molcel.2013.12.011
- Löffler AS, Alers S, Dieterle AM, Keppeler H, Franz-Wachtel M, Kundu M, Campbell DG, Wesselborg S, Alessi DR, Stork B. Ulk1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy 2011; 7:696-706; PMID:21460634; http://dx.doi.org/10.4161/auto.7.7.15451
- Bartholomeusz GA, Talpaz M, Kapuria V, Kong LY, Wang S, Estrov Z, Priebe W, Wu J, Donato NJ. Activation of a novel Bcr/Abl destruction pathway by WP1130 induces apoptosis of chronic myelogenous leukemia cells. Blood 2007; 109:3470-8; PMID:17202319; http://dx.doi.org/10.1182/blood-2006-02-005579
- Kapuria V, Levitzki A, Bornmann WG, Maxwell D, Priebe W, Sorenson RJ, Showalter HD, Talpaz M, Donato NJ. A novel small molecule deubiquitinase inhibitor blocks Jak2 signaling through Jak2 ubiquitination. Cell Signal 2011; 23:2076-85; PMID:21855629; http://dx.doi.org/10.1016/j.cellsig.2011.08.002
- Kapuria V, Peterson LF, Fang D, Bornmann WG, Talpaz M, Donato NJ. Deubiquitinase inhibition by small-molecule WP1130 triggers aggresome formation and tumor cell apoptosis. Cancer Res 2010; 70:9265-76.; PMID:21045142; http://dx.doi.org/10.1158/0008-5472.CAN-10-1530
- Sun H, Kapuria V, Peterson LF, Fang D, Bornmann WG, Bartholomeusz G, Talpaz M, Donato NJ. Bcr-Abl ubiquitination and Usp9x inhibition block kinase signaling and promote CML cell apoptosis. Blood 2011; 117:3151-62; PMID:21248063; http://dx.doi.org/10.1182/blood-2010-03-276477
- Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol 1998; 143:1883-98; PMID:9864362; http://dx.doi.org/10.1083/jcb.143.7.1883
- Alemu EA, Lamark T, Torgersen KM, Birgisdottir AB, Larsen KB, Jain A, Olsvik H, Overvatn A, Kirkin V, Johansen T. ATG8 family proteins act as scaffolds for assembly of the ULK complex: sequence requirements for LC3-interacting region (LIR) motifs. J Biol Chem 2012; 287:39275-90; PMID:23043107; http://dx.doi.org/10.1074/jbc.M112.378109
- Peddaboina C, Jupiter D, Fletcher S, Yap JL, Rai A, Tobin RP, Jiang W, Rascoe P, Rogers MK, Smythe WR, et al. The downregulation of Mcl-1 via USP9X inhibition sensitizes solid tumors to Bcl-xl inhibition. BMC Cancer 2012; 12:541; PMID:23171055; http://dx.doi.org/10.1186/1471-2407-12-541
- Peng Z, Maxwell DS, Sun D, Bhanu Prasad BA, Schuber PT, Jr., Pal A, Ying Y, Han D, Gao L, Wang S, et al. Degrasyn-like symmetrical compounds: Possible therapeutic agents for multiple myeloma (MM-I). Bioorg Med Chem 2014; 22:1450-8; PMID:24457091 ; http://dx.doi.org/10.1016/j.bmc.2013.12.048
- Wang S, Kollipara RK, Srivastava N, Li R, Ravindranathan P, Hernandez E, Freeman E, Humphries CG, Kapur P, Lotan Y, et al. Ablation of the oncogenic transcription factor ERG by deubiquitinase inhibition in prostate cancer. Proc Natl Acad Sci U S A 2014; 111(11):4251-6; PMID:24591637; http://dx.doi.org/10.1073/pnas.1322198111
- Harris DR, Mims A, Bunz F. Genetic disruption of USP9X sensitizes colorectal cancer cells to 5-fluorouracil. Cancer Biol Therapy 2012; 13:1319-24; PMID:22895071; http://dx.doi.org/10.4161/cbt.21792
- Xu P, Duong DM, Seyfried NT, Cheng D, Xie Y, Robert J, Rush J, Hochstrasser M, Finley D, Peng J. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 2009; 137:133-45; PMID:19345192; http://dx.doi.org/10.1016/j.cell.2009.01.041
- Kobayashi S, Yoneda-Kato N, Itahara N, Yoshida A, Kato JY. The COP1 E3-ligase interacts with FIP200, a key regulator of mammalian autophagy. BMC Biochem 2013; 14:1; PMID:23289756; http://dx.doi.org/10.1186/1471-2091-14-1
- Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol 2009; 10:550-63; PMID:19626045; http://dx.doi.org/10.1038/nrm2731
- Lazarus MB, Novotny CJ, Shokat KM. Structure of the Human Autophagy Initiating Kinase ULK1 in Complex with Potent Inhibitors. ACS Chem Biol 2015; 10(1):257-61; PMID:25551253; http://dx.doi.org/10.1021/cb500835z