
Abstract
In Saccharomyces cerevisiae, a constitutive biosynthetic transport pathway, termed the cytoplasm-to-vacuole targeting (Cvt) pathway, sequesters precursor aminopeptidase I (prApe1) dodecamers in the form of a large complex into a Cvt vesicle using autophagic machinery, targeting it into the vacuole (the yeast lysosome) where it is proteolytically processed into its mature form, Ape1, by removal of an amino-terminal 45-amino acid propeptide. prApe1 is thought to serve as a scaffolding cargo critical for the assembly of the Cvt vesicle by presenting the propeptide to mediate higher-ordered complex formation and autophagic receptor recognition. Here we report the X-ray crystal structure of Ape1 at 2.5 Å resolution and reveal its dodecameric architecture consisting of dimeric and trimeric units, which associate to form a large tetrahedron. The propeptide of prApe1 exhibits concentration-dependent oligomerization and forms a stable tetramer. Structure-based mutagenesis demonstrates that disruption of the inter-subunit interface prevents dodecameric assembly and vacuolar targeting in vivo despite the presence of the propeptide. Furthermore, by examining the vacuolar import of propeptide-fused exogenous protein assemblies with different quaternary structures, we found that 3-dimensional spatial distribution of propeptides presented by a scaffolding cargo is essential for the assembly of the Cvt vesicle for vacuolar delivery. This study describes a molecular framework for understanding the mechanism of Cvt or autophagosomal biogenesis in selective macroautophagy.
Abbreviations
| 5′-IAF | = | 5-iodoacetamidofluorescein |
| Ape1 | = | aminopeptidase I |
| Atg | = | autophagy-related |
| AUC | = | analytical ultracentrifugation |
| Cvt | = | cytoplasm-to-vacuole targeting |
| DDM | = | n-dodecyl-β-D-maltopyranoside |
| EM | = | electron microscopy |
| FM 4–64 | = | N-(3-triethylammoniumpropyl)-4-(6-(4-(diethylamino) phenyl) hexatrienyl) pyridinium dibromide |
| FP | = | fluorescence polarization |
| GFP | = | green fluorescent protein |
| MBP | = | maltose binding protein |
| Ni-NTA | = | nickel-nitrilotriacetic acid |
| PAS | = | phagophore assembly site |
| TEV | = | tobacco etch virus. |
Introduction
Selective macroautophagy (hereafter autophagy) plays a critical role in the clearance of defective aggregates and organelles that cause neurodegeneration or other diseases.Citation1,2 Many kinds of targeted cargos of selective autophagy have been described; they include mitochondria, peroxisomes, ribosomes, the endoplasmic reticulum, aggregated proteins, and pathogens.Citation3-6 The autophagic receptors recruit the autophagic machinery, which forms double-membrane vesicles known generally as autophagosomes (additional terms are sometimes used depending on the specific cargo, such as mitophagosomes, etc.), for cargo engulfment and subsequent delivery to the lysosome/vacuole. The autophagic machinery is not only employed in import of selected cargos to the lysosome/vacuole for degradation, but is also involved in important cellular pathways such as the secretion of cytokines, endosomal transport, as well as lysosomal trafficking and activation of hydrolytic enzymes by the cytoplasm-to-vacuole targeting (Cvt) pathway.Citation7-9 Therefore, it is essential to understand the structural mechanism of cargo recognition by the autophagic receptors and their role in autophagosome formation, which may provide insight into curing diseases by targeting the autophagic machinery.
In the yeast Saccharomyces cerevisiae, aminopeptidase I (Ape1) is a dodecameric chambered hydrolase involved in protein degradation in vacuoles (the yeast lysosomes). It is transported to vacuoles by the unique Cvt pathway under vegetative growth conditions and by the autophagy pathway during starvation.Citation10,11 Ape1 is synthesized in the cytosol as a precursor form, prApe1, which contains an N-terminal 45 amino acid propeptide.Citation12 It self-assembles into dodecamers, which then form higher-ordered structures, termed the Ape1 complex, in a propeptide-dependent manner.Citation13 The exposed propeptides are then recognized by the autophagic receptor autophagy-related (Atg) 19 (Atg19). Together with Atg11, the Atg19-bound Ape1 complex (now referred to as a Cvt complex) facilitates the formation of Cvt vesicles (an autophagosome-like compartment that selectively sequesters the Ape1 complex) and the organization of the phagophore assembly site (PAS).Citation14,15 The Cvt vesicle fuses with the vacuole and releases the inner membrane-bound cargo content into the vacuole, where it is now called a Cvt body.Citation16,17 Degradation of the inner membrane by the lipase Atg15 releases the prApe1 cargo and thus triggers the removal of the propeptide by the vacuole-resident Prb1/proteinase B to generate mature Ape1, which functions as an aminopeptidase in the vacuolar lumen.Citation18,19
The Cvt pathway in S. cerevisiae is the simplest and best characterized form of selective autophagy. The Cvt pathway recruits autophagic machinery around the targeted cargos facilitating assembly of lipid bilayers, shaping them into a double-membrane Cvt vesicle (140–160 nm) for delivery of its cargo to the vacuole.Citation20 However, the role of the spatial distribution of the ligands presented on a selected cargo recognized by autophagic receptors has not been investigated by structural analysis. Previously, it was thought that prApe1 serves as a scaffolding cargo involved in Cvt vesicle formation by employing the N-terminal propeptide, which acts as a ligand mediating Ape1 complex formation and autophagic receptor recognition, both of which are necessary for vacuolar delivery.Citation13 Yet, it is unclear how the propeptide may drive oligomerization of prApe1. Moreover, the structural role of prApe1 in presenting the propeptide to direct the formation of the Ape1 complex into an effective cargo for the Cvt pathway has remained unknown. Here, we present the crystal structure of Ape1 at 2.5 Å resolution, which reveals a dodecameric architecture organized by dimeric and trimeric units that associate to form a tetrahedron. Structure-based mutagenesis of prApe1 shows the essential roles of the tetrahedral assembly in presenting the propeptides for autophagic receptor recognition and vacuolar targeting in vivo. The combined cross-linking and analytical ultracentrifugation (AUC) analyses demonstrate that the propeptide forms a stable tetramer in solution. To gain further insight, we have constructed diverse propeptide-fused exogenous protein assemblies with various quaternary structures and distinct spatial presentation of the propeptides to examine their vacuolar delivery via the Cvt pathway in vivo. Our results suggest that the spatial distribution of the propeptides presented on a 3-dimensional scaffolding cargo is essential for proper assembly of the Cvt vesicle to ensure vacuolar delivery. Taken together, this work sheds light on the molecular assembly process fundamental to vesicle formation in the Cvt pathway during selective autophagy.
Results
Overall structure of Ape1
For the structural analysis of Ape1, we have expressed in bacterial cells recombinant full-length prApe1 and a truncated version corresponding to the mature form without the N-terminal propeptide region (residues Met1-Leu45), Ape1. Unlike Ape1, which expressed well, recombinant prApe1 was prone to aggregation; however, it could be solubilized with the detergent n-dodecyl-β-D-maltopyranoside (DDM). We focused on the Ape1 crystals and were able to solve the structure of Ape1 at 2.5 Å resolution. The X-ray structure of Ape1 was determined by molecular replacement (see Materials and Methods) and the final model was refined to Rwork/Rfree = 0.215/0.246. Residues Met1-Glu2, Pro165-Glu176, Ile185-Asn195 and Lys403-Gly409 were not visible due to absent electron density in the map. Data collection and structure refinement statistics are provided in . In the rhombohedral unit cell with 4 monomers per asymmetric unit, Ape1 assembles into the physiologically relevant dodecamer with tetrahedral arrangement of monomers contributed from 3 asymmetric units, which are related by 3-fold crystallographic symmetry. Each monomer contains 2 domains (), with the active site residues His88, Asp90, Asp259, Glu295, Glu296, Asp341, and His435, which are well conserved in the family M18 of metalloaminopeptidases, housed in a cleft at the domain interface (Fig. S1). The proteolytic domain (residues His3-Ala91 and Lys242-Leu470) consists of a large 8-stranded β-sheet with 9 flanking α-helices and a small sheet of 3 antiparallel β-strands abutted by a small helix (; Fig. S2A). Emerging from the proteolytic domain is the wing domain (residues Leu92-Gln241), which consists of an anti-parallel β-sheet flanked by several helices (). Distinct dimeric and trimeric units could be identified from the dodecameric tetrahedron of Ape1. The dimeric unit on each edge of the tetrahedron, related by 2-fold symmetry, is formed by association of the wing domain of a monomer subunit with the proteolytic domain of the paired subunit, and vice versa (). By contrast, the trimeric unit forms each vertex of the tetrahedron and is related by 3-fold symmetry, with the interface mediated by the proteolytic domains of the 3 contributing monomer subunits (). The edge of the tetrahedron is about 115 Å in length and each facet has a cavity of 26 Å in diameter and a 10 Å narrow opening at each vertex; similar architecture is found in other members of the family M18 proteases with known structures.Citation21,22 We were also able to characterize the structure of DDM-prApe1 by negative stain single-particle electron microscopy (EM). In the negatively stained specimen, DDM-prApe1 particles were well distributed and displayed a broad range of orientations (). The 3-dimensional reconstruction of DDM-prApe1, determined to 19 Å resolution (), showed a tetrahedron of 15 nm in diameter, with a hollow pocket in the center of the cage. The negative EM structure of DDM-prApe1 agrees well with the crystallographic dodecamer of Ape1, lending support to the physiological relevance of the latter (; Fig. S2B). The result also suggests that the propeptide does not affect the organization of the subunits observed from the crystal structure of Ape1. Note that because the DDM-prApe1 does not aggregate in solution, our presented EM structure could not show structural information on propeptide-mediated formation of the Ape1 complex. Future single-particle EM analysis of detergent-free prApe1 will be necessary to understand the process of Ape1 complex formation.
Figure 1. The structure of Ape1. (A) Each monomer of Ape1 consists of 2 domains: the wing domain (red) and the proteolytic domain (green). The dashed lines indicate missing residues in the crystal. (B) The dimeric unit forming the tetrahedron edge is shown in cylinder and tube models for each monomer, respectively. The 2-fold symmetry axis is perpendicular to the page plane. (C) The trimeric unit forming the tetrahedron vertex is shown in cylinder, tube, and loop models for each monomer, respectively. Corresponding schematic cartoons of the Ape1 assembly highlighting protomer interaction in the tetrahedron are shown below. (D) A close-up view of the intersubunit interface involving the proteolytic domain of Ape1. The residues of the proteolytic domain in the tube model selected for insertion mutagenesis and their interacting residues from the adjacent subunit are highlighted in yellow sticks.
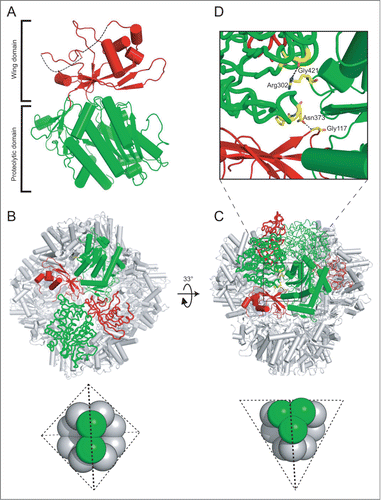
Figure 2. Single-particle reconstruction of DDM-solubilized prApe1. (A) Raw negative stain micrograph of DDM-prApe1. Scale bar: 100 nm. (B) Representative 197 class averages derived from about 30,000 particles. (C) Representative 198 projections derived from the reconstruction model. (D) Euler angle distribution plot of particles used in the reconstruction of DDM-prApe1 over an asymmetric unit. The position of each circle represents the orientations of particles belonging to the corresponding projection; the darkness of each circle is proportional to the relative number of views assigned for that projection. (E) The Fourier shell correlation (FSC) plot from refined density map of DDM-prApe1 is shown. The resolution of the final reconstruction was estimated to be 19 Å based on the 0.5 criterion of the FSC coefficient (dashed lines). (F) Three different views of the EM reconstruction shown in translucent isosurface with the fitted crystal structure of Ape1 shown in cyan ribbons. Figures were prepared using Chimera.
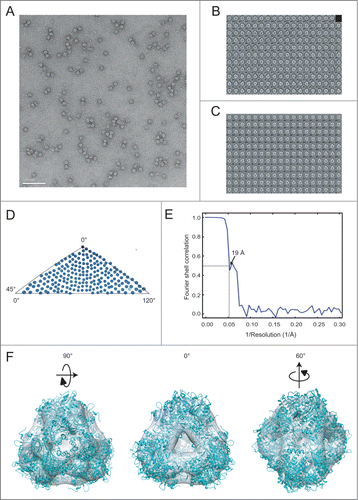
Table 1. Data collection and crystallographic refinement statistics
Propeptides mediate tetramerization of fusion proteins
The propeptides play a critical role in mediating the assembly of prApe1 complexes.Citation13 To further examine the oligomerization of the propeptide in vitro, we overexpressed and purified recombinant propeptide using E. coli cells and studied its biochemical properties. The sedimentation coefficient c(s) of 29 μM propeptide was ∼0.7S, which corresponded to the monomeric form. We carried out AUC measurements over the concentration range from 20 μM to 250 μM of propeptide, which exhibited concentration-dependent oligomerization because the absorbance of a population of ∼1.5S, corresponding to the size of tetramer, increased as a function of propeptide concentration (; Fig. S3). For quantitative analysis of the binding affinity, the isotherms of signal weighted-average sedimentation coefficient Sw values calculated from integration of c(s) between 0.5 S and 2 S were loaded into SEDPHAT for fitting.Citation23 The best fit was obtained by using the monomer-dimer-tetramer equilibrium model, and the Kd12 and Kd24 of 226 μM and 23 μM were determined (). The fit of the data with the monomer-dimer or the monomer-tetramer equilibrium models was not satisfactory (Fig. S4). It seems that the propeptide forms dimers with lower affinity, but dimerization effectively promotes stable tetramer formation. We also performed cross-linking experiments of propeptide at concentrations of 58.3 μM and 200 μM to analyze its oligomeric states, which showed that the amount of tetrameric propeptide accumulated as a function of time but did not further form species with higher molecular weight (). In order to examine whether the propeptide can direct the tetramerization of an exogenous protein, we fused the propeptide to the pyrin domain of NLRP10/NALP10/PYNOD, a monomeric protein-protein interaction domain.Citation24 The oligomeric state of the fusion protein, termed prPYNOD_PYD was assessed by using AUC. Comparing with wild-type PYNOD_PYD, with c(s) value of ∼1.6S, prPYNOD_PYD yielded c(s) of 1.94S and 5.25S, respectively, confirming that prPYNOD_PYD dimerizes and further oligomerizes into a tetramer (). Finally, we examined the aggregation of prApe1 conjugated with 5-iodoacetamidofluorescein (5′-IAF) in vitro using fluorescence polarization (FP). For this assay, a fusion protein of prApe1 was made with a cleavable N-terminal maltose-binding protein (MBP) tag. The highly soluble MBP tag enhanced the solubility of prApe1 such that MBP-prApe1 was expressed as a dodecameric form, judging by dynamic light scattering (Fig. S5A). Aggregation of 5′-IAF labeled MBP-prApe1 was initiated by addition of the tobacco etch virus (TEV) protease to remove the MBP tag from the fusion protein. We found that within 20 min of TEV treatment, most of the MBP tag was cleaved off from the fusion protein (Fig. S5B). Since MBP has no cysteine available for fluorescent probe labeling, the free tag does not contribute to the FP signal. The increase in FP, which indicates aggregation of prApe1, was monitored as a function of time. The result showed that prApe1 of various concentrations (5–20 μM) form a stable aggregate in 20 h, suggesting that free dodecameric prApe1 spontaneously aggregates in vitro ().
Figure 3. Oligomerization of the propeptide. (A) The propeptide shows concentration-dependent oligomerization. In the sedimentation coefficient distributions c(s) plots, the distributions were normalized with respect to various loading concentrations. (B) The resulting Sw isotherms from integration of c(s) for propeptides. In the isotherm plots, solid circles are the Sw data from the dilution series, and the solid line is the better-fit isotherm with a monomer-dimer-tetramer model. (C) SDS-PAGE analysis of the purified propeptide after cross-linking with glutaraldehyde. 58.3 μM (left) and 200 μM (right) of purified propeptide were cross-linked and the samples were run on a 15% polyacrylamide gel. S, SeeBlue® Plus2 protein ladder; B, Benchmark™ protein ladder; C, control without glutaraldehyde, in which the propeptide migrated as a monomer and an SDS-induced dimer presumably caused by hydrophobic interactions. Also note that the cross-linking treatment caused all crossed-linked species to migrate as a smaller apparent MW in SDS-PAGE, presumably due to formation of internal cross-links, which reduce the Stoke's radius. The positions of molecular mass marker proteins (in kDa) in the most left lanes are indicated. The reaction times are labeled on the top of the SDS-PAGE. (D) prPYNOD_PYD oligomerizes in a propeptide-dependent manner. (E) Fluorescence polarization (FP) assay of prApe1 aggregate formation. mP, unit for FP.
Tetrahedral shape is required for prApe1 import into the vacuole
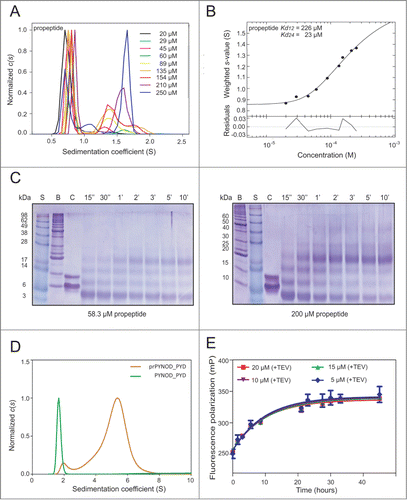
In order to examine whether the role of the tetrahedral assembly of the dodecameric prApe1 is crucial for its function, we performed structure-based mutagenesis to alter the oligomeric states of prApe1. We reasoned that due to the large buried surface area (at least 1800 Å2) at the oligomerization interfaces, introducing point mutations may be insufficient to disrupt the interfaces. Hence, in order to effectively do so without disturbing the tertiary structure of the Ape1 monomer, we chose to insert a 10-residue linker peptide (MSDYDIPTTE) in the proteolytic domain, into a loop between residues Gly421 and Ala422; this loop is buried at the interface between the proteolytic domains at the tetrahedron vertex (). The resulting mutant, termed Ape1-L1, is expected to prevent the formation of the vertex trimers required for tetrahedral assembly, by introducing steric hindrance. However, this insertion mutant should not prevent 2 of the vertex subunits from association; in other words, the L1 mutant may instead assemble into a hexamer with a structure similar but non-identical to the face of the Ape1 tetrahedron. In addition, another insertion to a different loop in the proteolytic domain was made in Ape1-L1 between the residues Pro372 and Asn373; the loop is critically involved in interaction of the proteolytic domain of a monomer subunit with the wing and proteolytic domains from a neighboring subunit. The resulting double insertion mutant, Ape1-L2, is expected to disrupt all subunit interaction involving the proteolytic domain observed in the tetrahedron assembly ().
Analytical ultracentrifugation was used to verify the altered oligomeric states of the 2 mutants. Sedimentation velocity studies showed that wild-type Ape1 had a sedimentation coefficient of 17.8S, corresponding to the dodecameric state. By contrast, Ape1-L1 shows a c(s) value of 12.2S, which corresponds to the hexameric state. Finally, Ape1-L2 consisted of 2 discrete species with 5.83S and 3.76S, representing the dimeric and monomeric states, respectively. The monomer and dimer species could be further characterized with fractional populations of 69.6% and 28.8%, respectively ().
Figure 4. Structure-based insertion mutagenesis of Ape1. (A) AUC sedimentation velocity data for Ape1-WT, Ape1-L1, and Ape1-L2. The obtained continuous size distributions are plotted against the S value, corrected for buffer density and viscosity at 20°C. Parameters used in calculating the sedimentation coefficients are viscosity = 0.01023 and density = 1.0052. Disruption of the subunit interface of Ape1-WT (red) results in formation of hexamer (Ape1-L1; green) and monomer/dimer (Ape1-L2; blue). (B) Defective vacuole localization of prApe1-L1 and L2 mutants suggests a tetrahedral structural requirement for forming the prApe1 aggregate to direct Cvt vesicle formation, which is required for vacuole targeting. FM 4–64 stains the vacuolar membrane. (C) Subcellular localization of GFP-fused prApe1-WT/L1/L2 co-transformed with RFP-Atg8 in yeast ape1Δ cells monitored by fluorescence microscopy after growing in nutrient-rich YPD medium (control) or SD-N medium (starvation) for 4 h.
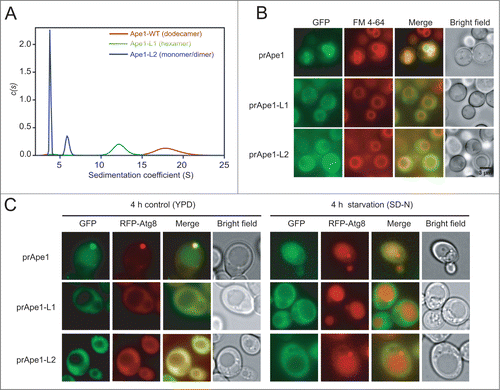
We then made the same L1 and L2 mutations in the yeast construct pRS416-GFP-prApre1 and transformed them into the yeast APE1-knockout strain (SEY6210 ape1Δ) and compared their subcellular localizations with that of prApe1-WT using fluorescence microscopy. As a positive control, the GFP signal of prApe1-WT was confined to the inside of vacuoles, whose membrane was labeled with a lipophilic styryl dye, N-(3-triethylammoniumpropyl)-4-(p-diethylamino phenylhexatrienyl)-pyridinium dibromide (FM 4–64), indicating that GFP-prApe1 formed complexes and was effectively delivered to the vacuole (; Fig. S6). By contrast, about 80% of GFP-prApe1-L1 cells showed no perivacuolar puncta and all of them presented a cytosolic GFP signal, suggesting a defective vacuolar delivery. Similarly, all of the GFP-prApe1-L2 resulted in diffuse cytosolic GFP fluorescence (). These results suggest that the hexameric and dimeric/monomeric forms of mutant prApe1 are defective in assembly into complexes, which is unlike prApe1-WT (see above), and are defective in vacuolar targeting as demonstrated by fluorescence microscopy. A previous study has shown that the Ape1 complex is not essential for autophagosome formation;Citation17 therefore, the cytoplasmic localization of the L1/L2 mutants is not likely due to defective autophagic flux in cells expressing the mutants. To verify this, we have found that ape1Δ yeast cells co-expressing GFP-prApe1-WT or -L1/L2 mutants with RFP-Atg8 indeed showed the expected localization of Atg8 at the PAS or in the cytoplasm under normal growth conditions, but in vacuoles during starvation ().
The propeptide of prApe1 interacts with Atg19.Citation13 To see whether the prApe1-L1 and -L2 mutants retain Atg19 binding ability, we prepared His6-tagged prApe1-L1 and prApe1-L2 to test their interaction with untagged Atg19 using Ni-NTA affinity isolation assays. The results show that these 2 mutants bind to Atg19 (Fig. S7). Together, these results suggest that in addition to the propeptide, mediating Atg19 interaction, a tetrahedral shape is also essential for prApe1 import into the vacuole by the autophagic machinery.
Propeptide-fused exogenous protein assemblies are imported into the vacuole
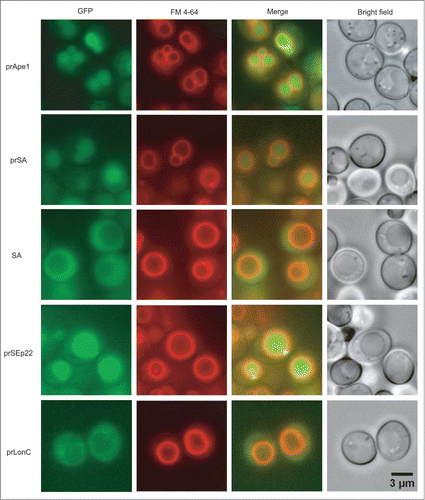
The above mutational analysis with the L1 and L2 mutants supports the idea that the presence of the propeptides, which are recognized by Atg19, is not sufficient to ensure proper assembly of the Cvt vesicle onto the Ape1 complex. This result suggests that the structure formed by the prApe1 dodecamer, but not hexamer, dimer, or monomer, may play an important role to present the propeptides for aggregation into a 3-dimensional scaffolding cargo necessary for proper assembly of the Cvt vesicles. There exist other non-prApe1 proteins, which form a similar dodecameric structure but lack a propeptide.Citation21 To understand whether the 3-dimensional spatial distribution of the propeptides on a dodecameric scaffold is essential for Cvt vesicle formation, we hypothesized that when fusing the propeptide to other non-prApe1 proteins capable of self-assembly into a prApe1-like dodecameric structure, these exogenous fusion proteins may be converted into effective Cvt cargos for vacuolar delivery. To test the hypothesis, we have identified 3 suitable cytosolic candidates with an exposed N terminus including Salmonella aminopeptidase SA, the bacterial DNA-binding protein SEp22, and the bacterial Lon-like protease MtaLonC.Citation25,26 Being a bacterial homolog of prApe1, SA also assembles into a tetrahedral dodecamer. Similarly, SEp22 forms a dodecameric shell. By contrast, MtaLonC forms a hexameric assembly. The genes of these exogenous proteins were cloned into the yeast centromere plasmid pRS416 and constructed into a fusion chimera with an N-terminal GFP-propeptide tag to visualize their complex formation and subcellular localization in vivo using fluorescence microscopy. Comparing to GFP-SA, whose GFP fluorescence was diffuse in the cytosol, the fluorescence signal of propeptide-fused SA (GFP-prSA) formed perivacuolar puncta and the signal was localized specifically inside the yeast vacuoles (; Fig. S6), indicating that GFP-prSA was transported to the vacuole in a propeptide-dependent manner. GFP-prSEp22-transformed yeast cells also showed both strong vacuolar staining and perivacuolar punctate structures, demonstrating that prSEp22 formed complexes and was delivered into the vacuole as well. In contrast, GFP-prLonC was localized only in the cytosol, with few perivacuolar puncta (; Fig. S6).
Figure 5. The aggregation and subcellular localization of propeptide-fused exogenous protein assemblies in yeast ape1Δ cells monitored by fluorescence microscopy. Propeptide fusion proteins are designated by the prefix “pr.” LonC denotes MtaLonC.
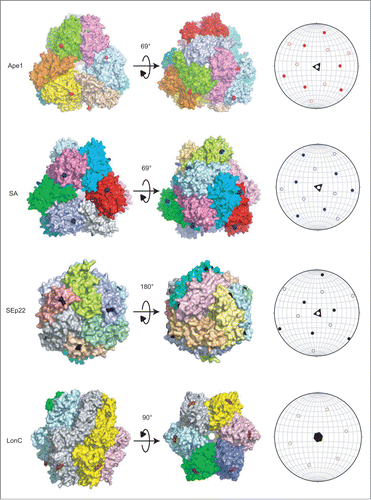
To help visualize the 3-dimensional distribution of the propeptides on the surface of the tested protein assemblies, we have made a stereographic projection-like plot for each of them. In the plot, a protein assembly particle with internal symmetry elements was placed inside of an imaginary sphere and the spherical coordinate system was used to define the positions of the residues at which the propeptides were fused (). The stereographic plots of Ape1, SA, and SEp22 showed projections, representing the exposed propeptides, derived from both sides of the hemispheres, which indicate that the propeptides have a uniform 3-dimensional spatial distribution on these protein assemblies. By contrast, the projection plot of MtaLonC shows only a 2-dimensional anisotropic distribution, suggesting that the propeptides fused to the N-termini of the protein assembly may only promote formation of layer-like oligomers. The resulting aggregate is expected to present the propeptides in a limited, 2-dimensional direction. Taken together, these results suggest that only if the propeptides (or autophagic ligands) are presented by a cargo with isotropic 3-dimensional spatial distribution, can the cargo then act like the Ape1 complex, which is effective in serving as a 3-dimensional scaffold for the assembly of the autophagosome-like vesicles such as the Cvt vesicle.
Figure 6. Spatial localization of the propeptides in Ape1 and the 3 exogenous protein assemblies in surface representations (left) as visualized by stereographic plots (right). The 4 proteins are shown in 2 orientations. Individual subunits are highlighted by distinct colors. The residues at which the propeptides are fused are colored as follows: Ape1 (red), SA (blue), SEp22 (black) and LonC (brown). In the stereographic plots, the solid and hollow patterns represent projection from upper and bottom poles, respectively.
Discussion
The Ape1 dodecamer is required for activity
Previous studies have shown that prApe1 dodecamers dissociate, under a pH lower than that in the vacuole, into catalytically inactive hexameric and dimeric forms.Citation27,28 Consistently, our engineered hexameric Ape1-L1 and dimeric L2 mutants are also enzymatically inactive. Our atomic structure of Ape1 reveals that the dodecamer is stabilized by extensive interaction between monomeric subunits. The buried area at subunit interfaces is approximately 1620–2517 Å2 and contributed by about 20 hydrogen bonds, which may explain why prApe1 oligomerizes into a dodecamer complex with a half-time of ∼2 min and is subsequently transported into the vacuole without disassembly.Citation29 The mutants Ape1-L1 and Ape1-L2 lacking peptidase activity confirm the importance of a self-compartmentalization strategy for efficient catalysis (Fig. S8A). The distribution of surface electrostatic potential of prApe1 may dictate the directional access of unfolded substrate polypeptide chains; here, in regard to the aminopeptidase, the negative charge surrounding the central cavity of Ape1 may attract the positively charged N terminus of the peptide (Fig. S8B).
Tetramerization of the propeptides may drive assembly of the dodecamers
Previous results showed that high concentrations of the propeptide could disassemble prApe1 aggregates in vitro, suggesting that the interaction between propeptides is weak.Citation13 Our AUC data confirmed the weak affinity between the propeptides, which was characterized by a dissociation constant (Kd) in the micromolar range. Although the structure of the propeptide is not available, this 45-amino-acid fragment is predicted to form a helix-turn-helix.Citation12 We calculated an in silico structural model of the propeptide by using the I-TASSER server, which gave the best model of the propeptide with a confidence score (C) of −1.6 (C scores typically range from −5 to 2; the higher the score is, the higher the confidence in the model) and a template modeling TM score of 0.52 (a TM score of >0.5 indicates a model of correct topology).Citation30 The model shows a helix-turn-helix structure as predicted, with a linker of 10 residues at the C terminus (Fig. S9A). The outer face of the first helix H1 is comprised of charged and polar residues and the hydrophobic residues are lining the inside. The outer face of the second helix H2 shows a negatively charged and hydrophobic surface. We fused the propeptide to the N terminus of our crystal structure to produce a theoretical model of full-length prApe1 (Fig. S9B). It is possible that the propeptides may lay on the face of the tetrahedron because there are positively charged patches near the N-terminal ends of Ape1 which may attract the second helix of the propeptide. However, this conformation would not be compatible with the dimerization-tetramerization results of our propeptide-fused protein analyses. A more likely alternative model is such that each propeptide protrudes from the tetrahedron face, mediating the clusters of propeptide of separate dodecamers to interact (Fig. S9B and C).
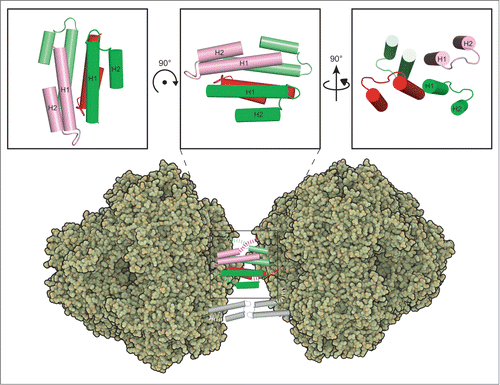
Intriguingly, the cross-linking, AUC, and chimera protein experiments all showed that propeptides form stable tetramers. Based on the prApe1 model, it is possible that 2 propeptides each presented from one tetrahedron face via a 10-residue linker may interact with 2 other propeptides from another tetrahedron face to form a stable tetramer (). Propeptide tetramer formation predicts that not all 3 of the propeptides presented from a tetrahedron face are required to mediate complex formation. Accordingly, a previous fluorescence microscopy study has shown that in wild-type yeast cells prApe1Δ2–45-GFP exhibits green punctate structures and green vacuoles, indicating that the aggregates may have formed from a mixture of prApe1 and prApe1Δ2–45 subunits; the latter lacking the propeptide.Citation13 This result indeed demonstrates that not all of the propeptides are required for complex formation. Site-directed mutagenesis and deletion experiments showed that the first predicted amphipathic α-helix and β-turn of the propeptides are important for prApe1 maturation and localization.Citation12 Hydrophobic interaction is important in prApe1 aggregation due to the stability of prApe1 aggregates in buffers that potentiate hydrophobicity.Citation13 We speculate that the nonpolar face of the H1 helix (Met1- Val20), which is rich in leucines (heptad pattern MEEQREILaEQLdKKTLaQMLdTV), is involved in forming the hydrophobic core of the tetramer. Previous studies have demonstrated that an α-helical peptide containing leucines at the a and d positions can form a stable tetrameric helical bundle.Citation31 Mutations of these 4 residues shows complete inhibition in prApe1 processing; deletion or insertion in this region also disrupts the hydrophobic periodicity of the propeptide, which leads to defective prApe1 processing and delivery.Citation12,13 Future structural analysis will be required to further confirm the proposed tetrameric model of the propeptides.
Figure 7. Proposed model for prApe1 complex assembly. Two propeptides (colored in red and pink, respectivey) presented from one tetrahedron face may interact with 2 other propeptides (colored in green and lime, respectively) from another tetrahedron face to form a stable tetramer. The upper panel represents 3 views of a propeptide tetramer modeled by superimposing the H1 helices of 4 I-TASSER models (see text) based on the crystal structure of a 4-helical bundle (PDB code 4HB1). The structure of the propeptide tetramer has not been determined experimentally. The dashed lines indicate the linkers at the C terminus. The protein model figures of Ape1 were generated with QuteMol. Citation45
Isotropic 3-dimensional spatial distribution of the propeptides is crucial for Cvt vesicle formation
In ape1Δ cells, propeptide-fused SA and SEp22 can form aggregates and be delivered into vacuoles. This result demonstrates that the propeptide is a unique structural determinant for mediating aggregation of protein assemblies into an effective cargo of the Cvt pathway by recruiting the receptor Atg19 and other components of the autophagic machinery. SA and SEp22 form tetrahedral and spherical structures, respectively. Although the shapes of these protein assemblies are different, they are sufficient to present the fused propeptides effectively so as to ensure Cvt vesicle formation. Our work suggests that an isotropic 3-dimensional spatial distribution of the propeptides (serving as the ligands for autophagic receptors like Atg19) permitted on these 2 different structures is the key factor to enlarge the size of the cargo in 3 dimensions through assembly while allowing the autophagic receptor and adaptor proteins to recruit the phagophore membrane fragments around the cargo for effective engulfment. A recent study has shown that binding of the prApe1 cargo to Atg19 exposes multiple Atg8-binding sites of the latter, which improves the interaction of Atg19 with the Atg8 family proteins residing on the phagophore membrane.Citation32 The induced activity of prApe1 cargo-activated Atg19 to engage interaction with multiple Atg8 proteins on a phagophore membrane is thought to contribute to bending of the phagophore membrane around the cargo. Based on our results, isotropically distributed autophagic ligands, such as the propeptides of prApe1, presented on the outer shell of the cargo may further contribute to tight wrapping of the phagophore membrane around the cargo in selective autophagy. How binding of the prApe1 cargo induces the conformational change of Atg19 required for a subsequent membrane recruiting step awaits future structural studies.
Materials and Methods
Expression and purification of proteins
The sequences of full-length prApe1 (Met1-Leu514), Ape1 (Met1-Leu470), Atg19 (Met1- Leu415) and the propeptide (Met1-Leu45) were cloned into pETM11 (EMBO collection) and modified pET28a (Novagen, 69864–3) vectors respectively; all of the cloned genes contain an N-terminal 6xHis tag followed by a TEV cleavage site. The construct of Ape1-L1 was made by inserting a 10-residue linker (MSDYDIPTTE) between Gly421 and Ala42. Ape1-L2 was constructed by inserting the 10-residue linker in Ape1-L1 between Pro372 and Asn373. The prApe1-L1 and prApe1-L2 constructs were cloned in the same way as Ape1-L1 and Ape1-L2 but using prApe1 as template. The proteins were expressed in E. coli BL21(DE3) (BioAspect, BA-RH217-J40) and cells were cultured to an A600 of 0.7 and induced with 1 mM isopropyl β-D-thiogalactopyranoside (BioShop, IPT001) at 16°C overnight. Cell pellets were resuspended in lysis buffer (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 20 mM imidazole and Roche EDTA-free protease inhibitor cocktail [Roche, 04693116001]) and then ruptured by a high-pressure homogenizer (Avestin, EmulsiFlex-C3). After centrifugation at 35,000 × g at 4°C for 45 min, the supernatant fraction was applied to a nickel-nitrilotriacetic acid (Ni-NTA; Qiagen, 30230) column and washed with 50–100 mM imidazole (Sigma, 56750). The protein fraction, eluted with 250 mM imidazole, was digested with TEV protease (Life Technologies, 12575–015) to remove the N-terminal 6xHis tag and purified by a Superdex 200 column (GE Healthcare, 17–5175–01) equilibrated in 20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 5 mM DTT. Protein purity was analyzed by SDS-PAGE. Purified Ape1 was concentrated to 8 mg ml−1 for crystallization. The expression and purification procedure of prApe1-L1 and prApe1-L2 were performed as described above except the removal of the N-terminal His6-tag.
For MBP-fused prApe1, the full-length prApe1 sequence was cloned into pETM41 (EMBO collection). The expression of the MBP fusion protein in E. coli BL21(DE3) was induced with 1 mM isopropyl β-D-thiogalactopyranoside at 20°C for 4 h. After cell disruption and centrifugation at 35,000 × g at 4°C for 45 min, the supernatant fraction was incubated with Ni-NTA resin for 2 h. The resin was washed with 20 mM and 50 mM imidazole before elution with 250 mM imidazole. The eluted protein fraction was further purified by a Superose 6 (GE Healthcare, 17-5172-01) column equilibrated in 20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 5 mM DTT.
Crystallization and structure determination of Ape1
Crystals of Ape1 were obtained by mixing the protein solution with an equal amount of well reservoir solution consisting of 100 mM Tris-HCl, pH 7.2, 42.5% polyethylene glycol 400 (Hampton, HR2603), 0.1 M MgCl2, 1.1 M NaCl using the sitting-drop vapor diffusion method.Citation33 The crystal that gave the best X-ray diffraction to 2.5 Å was used for data collection of a total of 360 frames with 0.5° oscillation per frame. The diffraction data for Ape1 was collected on NSRRC beamline 15A (Taiwan). All images were processed using the HKL-2000 package.Citation34 Ape1 crystals belonged to space group R3 (). The structure was solved by molecular replacement using a monomer of the poly-alanine model of human aspartyl aminopeptidase generated by CHAINSAW as the search model (PDB code 4DYO),Citation35 which shares 30% sequence identity, using PHASER in CCP4 suite,Citation36,37 which gave a unique solution with TFZ = 11.0 and LLG = 387. Rigid-body refinement of the solution, which contained all 4 monomers of the search model as expected in the asymmetric unit, gave an Rfree of 0.43. After autobuilding using Buccaneer,Citation38 the Rwork and Rfree values were 0.224 and 0.280, respectively. Subsequently, the structure was manually rebuilt using Coot and refined using Refmac5.Citation39,40 All protein model figures were generated with PyMOL (v.1.1; Schrödinger). The atomic coordinates and structure factors for Ape1 were deposited in the Protein Data Bank (http://www.rcsb.org/) with the accession number 4R8F.
Yeast plasmid construction
The plasmid pRS416-CuGFP-prApe1 (a gift from Dr. Wei-Pang Huang, NTU, Taiwan) was the starting vector for making GFP-tagged constructs for prApe1-L1, prApe1-L2, SA, prSA, prSEp22, and prLonC. The DNA sequence encoding the linker MSDYDIPTTE was inserted after Gly465 of pRS416-CuGFP-prApe1 for prApe1-L1, and after Pro416 for prApe1-L2. For constructing prSA, prSEp22, and prLonC, the Nhe1 (for prSA and prSEp22 ) or BglII (for prLonC) restriction sites were introduced after the codon of Leu45, which is the C-terminal residue of the propeptide. The DNA fragment encoding SA and SEp22, containing flanking NheI sites, were amplified by PCR from the genomic DNA of Salmonella enterica subsp. Arizonae. The LonC gene fragment with flanking BglII sites was amplified by PCR from a plasmid published previously.Citation25 For making the construct SA, which is devoid of the propeptide, 2 SphI sites were introduced before Met1 and after Leu45 of pRS416-GFP-prSA, and the coding sequence of the propeptide was removed by cleavage with SphI prior to self-ligation of the digested plasmid.
Yeast transformation and FM 4-64 staining
The yeast strain used in this study was SEY6210 (MATα his3-Δ200 leu2-3,112 lys2-801 trp1-Δ901ura3-52 suc2-Δ9 GAL ape1Δ::LEU2 in SEY6210; a gift from Dr. Wei-Pang Huang). YPD medium was used for growing yeast cells. For nitrogen starvation, cells were incubated in nitrogen starvation (SD-N) medium (0.17% yeast nitrogen base without ammonium sulfate, or amino acids and containing 2% glucose). Yeast transformations were performed using the Alkali-Cation Yeast Transformation Kit (MP Biomedicals, 2200–200). For FM 4-64 (Molecular Probes, T13320) labeling, cells were incubated with 8 μM dye in YPD for 30 min, then washed with fresh medium and chased 60 min. Cells were then collected, resuspended in distilled water, and mounted on slides for microscope imaging using epifluorescence microscopy (Olympus BX61). The experiments were repeated 3 times and counted at least 400 cells.
For autophagic flux experiments, pRS414-RFP-Atg8 (a gift from Dr. Wei-Pang Huang) was cotransformed with pRS416-GFP-prApe1-WT and pRS416-GFP-prApe1-L1, or pRS416-GFP-prApe1-L2, using the same method as described above. Cells were grown in YPD medium overnight and then diluted in the same medium and further grown until OD600 = 1.0. Then the cells were collected by centrifugation and resuspended in SD-N medium to induce autophagy. After 4 h, the cells were imaged by fluorescence microscopy.
Analytical ultracentrifugation (AUC)
AUC experiments were carried out using an XL-A analytical ultracentrifuge (Beckman Coulter). Sedimentation velocity measurements using absorbance optics of reference buffer and samples of Ape1, Ape1-L1 and Ape1-L2 in 20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 5 mM DTT were performed at 20°C at a speed of 72,576-201,600 × g; and a concentration gradient from 20 μM to 250 μM of propeptide in phosphate-buffered saline (PBS; Sigma, P4417), 2 mM DTT was obtained at 290,304 × g. In the case of PYNOD_PYD and prPYNOD_PYD, the experiments were carried out at 201,600 × g in PBS, 5 mM DTT. The buffer density and viscosity were analyzed using the software SEDNTERP.Citation41 Data were fitted globally to a continuous c(s) distribution using SEDFIT software.Citation42 The summary of analytical ultracentrifugation data is listed in Table S1.
To analyze the binding affinity of the propeptide, the integration of c(s) distribution was performed to determine the isotherm of signal weighted-average sedimentation coefficient (Sw) as a function of protein concentration using the software GUSSI (http://biophysics.swmed.edu/MBR/software.html), kindly provided by Chad Brautigam (UTSW). The Sw isotherm was further loaded into SEDPHAT for fitting with the models.Citation23
Fluorescence polarization
MBP-prApe1 was dialyzed into PBS with 5 mM DTT and incubated with a 5-fold molar ratio of 5-iodoacetamidofluorescein to protein at 4°C overnight to label protein with the probe for recording the change of fluorescence polarization induced by self-aggregation. The mixture was purified in buffer (20 mM Tris-HCl, pH 7.4, 100 mM NaCl, 2 mM DTT) to remove free dye. 0.25 μM labeled MBP-prApe1 was incubated with 5, 10, 15, or 20 μM unlabeled proteins and aggregation was initiated by adding a 1:10 molar ratio of TEV protease to protein to remove His-MBP tag and monitor FP as a function of time using Paradigm (Beckman). The fluorescence polarization was calculated using the formula FP = ( I∥ − I⊥) / ( I∥ + I⊥), where I∥ and I⊥ are the intensity measured of the emission signal made parallel or orthogonal to the direction of polarizer. The experiments were repeated 3 times.
Peptidase activity assay
Peptidase activity assays were performed using L-leucine p-nitroanilide (Sigma, L9125) as the substrate. The reaction mixture contains 50 mM MES (Sigma, M2933), pH 6.0, 1 mM ZnCl2, 135 nM enzymes and 1 mM substrates and the measurement was carried out by monitoring OD405.
Cross-linking of the propeptide
A final concentration of 0.1% glutaraldehyde was added into various concentrations of the propeptide in PBS with 2 mM DTT at room temperature and then incubated for various time points and the reaction was terminated by adding 1M Tris-HCl, pH8.0. All of the samples were analyzed by 15% SDS-PAGE.
Dynamic light scattering
The average size and size distribution of the 3 μM MBP-prApe1 were measured by dynamic light scattering using a Zetasizer, Nano-ZS instrument (Malvern Instruments Ltd, UK). The intensity of the scattered light was detected at 90° to an incident beam and the results were analyzed using the automatic mode. Determined size was presented as the average value of 20 runs, with triplicate measurements within each run.
Stereographic projection plots
The coordinates of the centroid of a protein were identified using the Center of Mass Calculator server (http://sebastianraschka.com/Webapps/pdb_center_of_mass.html) based on PDB coordinates. Using Cartesian coordinates of the identified centroids, the coordinates of the CA atoms of the first residues C-terminal to the fused propeptides were then converted into polar coordinates with respect to the centroid of the protein using the formulae r = (x2+y2+z2) 1/2 (r is the distance between the atom CA and the centroid, x, y, z are the shifted Cartesian coordinates of CA after the centroid is set as the origin), ρ = cos −1[(x2+y2) 1/2/r] (ρ is the angle between the highest-fold internal symmetry axis and the CA atom, measured downward from the North pole of the sphere) and θ= tan−1 (y/x) (θ represents the angle measured in the horizontal plane). The sign value of z determines whether the locations of propeptide-fusion residues posit the North/upper (positive) or South/bottom (negative) poles, respectively. The stereographic projection plots were drawn using Stereonet 9 (by Rick Allmendinger) based on the trend (ρ), plunge (θ), and the symmetry operators.
Ni-NTA affinity isolation assay
2.4 μM of His6-tagged prApe1-L1 or prApe1-L2 was incubated with 7.2 μM untagged Atg19 in 100 μL of binding buffer containing 50 mM Tris-HCl, pH 8.0, 100 mM NaCl for 1 h on ice, followed by the addition of 100 μL Ni-NTA agarose beads (Qiagen, 30230) with end-to-end-rotation for 1 h at 4°C. The resin was collected by centrifugation at 345 × g for 5 min and then washed twice with 300 μL binding buffer. The bound protein was eluted with binding buffer containing 250 mM imidazole. Samples were analyzed by SDS-PAGE.
Electron microscopy, image processing, and 3-dimensional reconstruction
Four μl of freshly purified prApe1 at concentration of 20 μg ml−1 was adsorbed to a carbon-coated electron microscopy grid (Electron Microscopy Sciences, FCF200-CU) for 1 min and then stained with 2.5% uranyl acetate. Micrographs were obtained using a Tecnai F20 TWIN transmission electron microscope operating at 200 keV using a dose of (20e−/Å2) on a Gatan 4 k × 4 k CCD at a magnification of 62,000 (1.65 Å/pixel) and defocus values ranging from −1.6 to −3 μm. Single particle reconstruction of prApe1 was performed using EMAN2 software package.Citation43 Approximately 30,000 particles were selected using a semiautomatic particle selection procedure with the EMAN e2boxer and extracted into 128 ×128 pixel boxes. The images were CTF-corrected with EMAN2. 2D reference-free classification was performed exclusively within EMAN2 through iterative rounds of multivariate statistical analysis and multi-reference alignment. Based on the rotational symmetry observed in the eigenimages, tetrahedral symmetry was applied for the reconstruction and refinement. An initial model was built with class averages corresponding to top and side views and then refined in every dataset using iterative rounds of angular reconstitution. Refinement was done to remove images with high noise and artifacts. Each of the DDM-prApe1 image-class-averages was then compared to the projections from the reconstructed 3D volume. The images belonging to the class-averages with low radial profile correlation (less than 0.2) were rejected from the data set. The Fourier shell correlation of the EM map was calculated by EMAN2. The final map had a resolution of 19 Å based on the Fourier shell correlation with a cutoff value of 0.5. Three-dimensional EM map visualization and fitting of the crystal structures were produced using the fit-in-map module in Chimera.Citation44 The EM map of DDM-prApe1 has been deposited in the Electron Microscopy Data Bank (EMDB) (accession number EMD-6265).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
1067363_supplemental_files.zip
Download Zip (17.1 MB)Acknowledgments
We thank Dr. Wei-Pang Huang for his generous gifts of plasmids and yeast strain, and Jackie Yen for plasmid cloning. We are grateful to Drs. Chia-Yu Lu, Mao-Hua Deng, and Hong-Yi Lin at the Department of Geosciences of National Taiwan University for their valuable assistance and suggestions on the stereographic plots. We also thank Justine Huang and Chris Chang for suggestions on the manuscript. We acknowledge the staff at the 15A beamline of the National Synchrotron Radiation Research Center (NSRRC), Taiwan for their support on data collection. We acknowledge the use of the instruments in the Biophysics Core Facility at Academia Sinica.
Funding
This work was supported by Academia Sinica (to C.-I C. and G.-C.C.).
Related Research Data
References
- Lunemann JD, Schmidt J, Schmid D, Barthel K, Wrede A, Dalakas MC, Münz C. Beta-amyloid is a substrate of autophagy in sporadic inclusion body myositis. Ann Neurol 2007; 61:476-83; PMID:17469125; http://dx.doi.org/10.1002/ana.21115
- Dikic I, Johansen T, Kirkin V. Selective autophagy in cancer development and therapy. Cancer Res 2010; 70:3431-4; PMID:20424122; http://dx.doi.org/10.1158/0008-5472.CAN-09-4027
- Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol 2014; 16:495-501; PMID:24875736; http://dx.doi.org/10.1038/ncb2979
- Meijer WH, van der Klei IJ, Veenhuis M, Kiel JA. ATG genes involved in non-selective autophagy are conserved from yeast to man, but the selective Cvt and pexophagy pathways also require organism-specific genes. Autophagy 2007; 3:106-16; PMID:17204848; http://dx.doi.org/10.4161/auto.3595
- Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature 2010; 466:68-76; PMID:20562859; http://dx.doi.org/10.1038/nature09204
- Kirkin V, Lamark T, Sou YS, Bjorkoy G, Nunn JL, Bruun JA, Shvets E, McEwan DG, Clausen TH, Wild P, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell 2009; 33:505-16; PMID:19250911; http://dx.doi.org/10.1016/j.molcel.2009.01.020
- Yordy B, Tal MC, Hayashi K, Arojo O, Iwasaki A. Autophagy and selective deployment of Atg proteins in antiviral defense. Int Immunol 2013; 25:1-10; PMID:23042773; http://dx.doi.org/10.1093/intimm/dxs101
- Khalfan WA, Klionsky DJ. Molecular machinery required for autophagy and the cytoplasm to vacuole targeting (Cvt) pathway in S. cerevisiae. Curr Opin Cell Biol 2002; 14:468-75; PMID:12383798; http://dx.doi.org/10.1016/S0955-0674(02)00343-5
- Ruck A, Attonito J, Garces KT, Nunez L, Palmisano NJ, Rubel Z, Bai Z, Nguyen KC, Sun L, Grant BD, et al. The Atg6/Vps30/Beclin 1 ortholog BEC-1 mediates endocytic retrograde transport in addition to autophagy in C. elegans. Autophagy 2011; 7:386-400; PMID:21183797; http://dx.doi.org/10.4161/auto.7.4.14391
- Baba M, Osumi M, Scott SV, Klionsky DJ, Ohsumi Y. Two distinct pathways for targeting proteins from the cytoplasm to the vacuole/lysosome. J Cell Biol 1997; 139:1687-95; PMID:9412464; http://dx.doi.org/10.1083/jcb.139.7.1687
- Scott SV, Baba M, Ohsumi Y, Klionsky DJ. Aminopeptidase I is targeted to the vacuole by a nonclassical vesicular mechanism. J Cell Biol 1997; 138:37-44; PMID:9214379; http://dx.doi.org/10.1083/jcb.138.1.37
- Oda MN, Scott SV, Hefner-Gravink A, Caffarelli AD, Klionsky DJ. Identification of a cytoplasm to vacuole targeting determinant in aminopeptidase I. J Cell Biol 1996; 132:999-1010; PMID:8601598; http://dx.doi.org/10.1083/jcb.132.6.999
- Morales Quinones M, Winston JT, Stromhaug PE. Propeptide of aminopeptidase 1 protein mediates aggregation and vesicle formation in cytoplasm-to-vacuole targeting pathway. J Biol Chem 2012; 287:10121-33; PMID:22123825; http://dx.doi.org/10.1074/jbc.M111.311696
- Yorimitsu T, Klionsky DJ. Atg11 links cargo to the vesicle-forming machinery in the cytoplasm to vacuole targeting pathway. Mol Biol Cell 2005; 16:1593-605; PMID:15659643; http://dx.doi.org/10.1091/mbc.E04-11-1035
- Scott SV, Guan J, Hutchins MU, Kim J, Klionsky DJ. Cvt19 is a receptor for the cytoplasm-to-vacuole targeting pathway. Mol Cell 2001; 7:1131-41; PMID:11430817; http://dx.doi.org/10.1016/S1097-2765(01)00263-5
- Suzuki K, Kirisako T, Kamada Y, Mizushima N, Noda T, Ohsumi Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J 2001; 20:5971-81; PMID:11689437; http://dx.doi.org/10.1093/emboj/20.21.5971
- Suzuki K, Kamada Y, Ohsumi Y. Studies of cargo delivery to the vacuole mediated by autophagosomes in Saccharomyces cerevisiae. Dev Cell 2002; 3:815-24; PMID:12479807; http://dx.doi.org/10.1016/S1534-5807(02)00359-3
- Klionsky DJ, Cueva R, Yaver DS. Aminopeptidase I of Saccharomyces cerevisiae is localized to the vacuole independent of the secretory pathway. J Cell Biol 1992; 119:287-99; PMID:1400574; http://dx.doi.org/10.1083/jcb.119.2.287
- Teter SA, Eggerton KP, Scott SV, Kim J, Fischer AM, Klionsky DJ. Degradation of lipid vesicles in the yeast vacuole requires function of Cvt17, a putative lipase. J Biol Chem 2001; 276:2083-7; PMID:11085977; http://dx.doi.org/10.1074/jbc.C000739200
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12:814-22; PMID:20811353; http://dx.doi.org/10.1038/ncb0910-814
- Chaikuad A, Pilka ES, De Riso A, von Delft F, Kavanagh KL, Venien-Bryan C, Oppermann U, Yue WW. Structure of human aspartyl aminopeptidase complexed with substrate analogue: insight into catalytic mechanism, substrate specificity and M18 peptidase family. BMC Struct Biol 2012; 12:14; PMID:22720794; http://dx.doi.org/10.1186/1472-6807-12-14
- Sivaraman KK, Oellig CA, Huynh K, Atkinson SC, Poreba M, Perugini MA, Trenholme KR, Gardiner DL, Salvesen G, Drag M, et al. X-ray crystal structure and specificity of the Plasmodium falciparum malaria aminopeptidase PfM18AAP. J Mol Biol 2012; 422:495-507; PMID:22709581; http://dx.doi.org/10.1016/j.jmb.2012.06.006
- Schuck P. On the analysis of protein self-association by sedimentation velocity analytical ultracentrifugation. Anal Biochem 2003; 320:104-24; PMID:12895474; http://dx.doi.org/10.1016/S0003-2697(03)00289-6
- Su MY, Kuo CI, Chang CF, Chang CI. Three-dimensional structure of human NLRP10/PYNOD pyrin domain reveals a homotypic interaction site distinct from its mouse homologue. PloS one 2013; 8:e67843; PMID:23861819; http://dx.doi.org/10.1371/journal.pone.0067843
- Liao JH, Ihara K, Kuo CI, Huang KF, Wakatsuki S, Wu SH, Chang CI. Structures of an ATP-independent Lon-like protease and its complexes with covalent inhibitors. Acta Crystallogr D Biol Crystallogr 2013; 69:1395-402; PMID:23897463; http://dx.doi.org/10.1107/S0907444913008214
- Miyamoto T, Asahina Y, Miyazaki S, Shimizu H, Ohto U, Noguchi S, Satow Y. Structures of the SEp22 dodecamer, a Dps-like protein from Salmonella enterica subsp enterica serovar Enteritidis. Acta Crystallogr Sect F Struct Biol Cryst Commun 2011; 67:17-22; PMID:21206015; http://dx.doi.org/10.1107/S1744309110043113
- Marx R, Metz G, Rohn KH. The quaternary structure of yeast aminopeptidase I. Two. Geometric arrangement of subunits. Z Naturforsch C 1977; 32:938-43; PMID:146356
- Metz G, Marx R, Rohm KH. The quaternary structure of yeast aminopeptidase I. One. Molecular forms and subunit size. Z Naturforsch C 1977; 32:929-37; PMID:146355
- Kim J, Scott SV, Oda MN, Klionsky DJ. Transport of a large oligomeric protein by the cytoplasm to vacuole protein targeting pathway. J Cell Biol 1997; 137:609-18; PMID:9151668; http://dx.doi.org/10.1083/jcb.137.3.609
- Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 2008; 9:40; PMID:18215316; http://dx.doi.org/10.1186/1471-2105-9-40
- Apostolovic B, Danial M, Klok HA. Coiled coils: attractive protein folding motifs for the fabrication of self-assembled, responsive and bioactive materials. Chem Soc Rev 2010; 39:3541-75; PMID:20676430; http://dx.doi.org/10.1039/b914339b
- Sawa-Makarska J, Abert C, Romanov J, Zens B, Ibiricu I, Martens S. Cargo binding to Atg19 unmasks additional Atg8 binding sites to mediate membrane-cargo apposition during selective autophagy. Nat Cell Biol 2014; 16:425-33; PMID:24705553; http://dx.doi.org/10.1038/ncb2935
- Ducruix A, Giegé R. Crystallization of Nucleic Acids and Proteins: A Practical Approach. Oxford:IRL Press Oxford University Press 1992:82-90.
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography, Pt A 1997; 276:307-26; http://dx.doi.org/10.1016/S0076-6879(97)76066-X
- Stein N. CHAINSAW: a program for mutating pdb files used as templates in molecular replacement. J Appl Crystallogr 2008; 41:641-3; http://dx.doi.org/10.1107/S0021889808006985
- Mccoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr 2007; 40:658-74; PMID:19461840; http://dx.doi.org/10.1107/S0021889807021206
- Bailey S. The Ccp4 Suite - Programs for Protein Crystallography. Acta Crystallogr D 1994; 50:760-3; PMID:15299374; http://dx.doi.org/10.1107/S0907444993011898
- Cowtan K. The Buccaneer software for automated model building. One. Tracing protein chains. Acta Crystallogr D 2006; 62:1002-11; PMID:16929101; http://dx.doi.org/10.1107/S0907444906022116
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 2004; 60:2126-32; PMID:15572765; http://dx.doi.org/10.1107/S0907444904019158
- Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 2011; 67:355-67; PMID:21460454; http://dx.doi.org/10.1107/S0907444911001314
- Hayes D. LT, Philo J. Program Sednterp: Sedimentation Interpretation Program, Alliance Protein Laboratories, Thousand Oaks, CA. 1995.
- Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J 2000; 78:1606-19; PMID:10692345; http://dx.doi.org/10.1016/S0006-3495(00)76713-0
- Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol 2007; 157:38-46; PMID:16859925; http://dx.doi.org/10.1016/j.jsb.2006.05.009
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera-a visualization system for exploratory research and analysis. J Comput Chem 2004; 25:1605-12; PMID:15264254; http://dx.doi.org/10.1002/jcc.20084
- Tarini M, Cignoni P, Montani C. Ambient occlusion and edge cueing to enhance real time molecular visualization. IEEE Trans Vis Comput Graphics 2006; 12:1237-44; PMID:17080857; http://dx.doi.org/10.1109/TVCG.2006.115
- Weiss MS. Global indicators of X-ray data quality. J Appl Crystallogr 2001; 34:130-5; http://dx.doi.org/10.1107/S0021889800018227
- Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science 2012; 336:1030-3; PMID:22628654; http://dx.doi.org/10.1126/science.1218231