
Abstract
Cancer and diabetes are 2 multifactorial chronic diseases with tremendous impact on health worldwide. Metabolic risk factors play a critical role in fueling a wide range of cancers, but with undefined mechanisms. We recently reported that TRIB3, a stress-induced protein, mediates a reciprocal antagonism between autophagic and proteasomal degradation systems and connects insulin-IGF1 to malignant promotion. We found that several human cancer tissues express higher TRIB3 and phosphorylated IRS1 (insulin receptor substrate 1), which correlates negatively with patient prognosis. Silencing of TRIB3 not only restores insulin-IGF1-suppressed autophagic flux, but also attenuates tumor growth and metastasis. TRIB3 physically interacts with the autophagic receptor SQSTM1, and this interaction hinders the binding of SQSTM1 to LC3 and ubiquitinated proteins, leading to SQSTM1 accumulation and clearance inhibition of ubiquitinated proteins. Interrupting the TRIB3-SQSTM1 interaction with an α-helical peptide derived from SQSTM1 attenuates tumor growth and metastasis through activating autophagic flux. Our findings indicate that TRIB3 links insulin-IGF1 to cancer development and progression through interacting with SQSTM1. Thus, interrupting the TRIB3-SQSTM1 interaction may provide a potential strategy against cancers in patients with diabetes.
Type 2 diabetes mellitus (T2D) is a serious and growing health problem worldwide. Clinical and epidemiological studies have shown that diabetes carries an increased risk for a number of different forms of cancer. However, different medications against T2D result in entirely different outcomes in cancer development depending on their insulin-increasing or insulin-lowering efficacy. Diabetics treated with metformin have a significant decrease in cancer incidence than those who take insulin as therapy, or sulfonylurea drugs that increase insulin secretion from the pancreas. Cancer and T2D share a number of risk factors, such as obesity, age, lifestyle factors, and diet. These diseases also share some metabolic changes, among which high insulin and insulin-like hormones in circulation have been considered to be the most potential biological links between the 2 diseases, but the underlined molecular mechanisms are far from understood.
TRIB3 is one member of mammalian Tribbles homologs, which contain a Ser/Thr protein kinase-like domain but lack the ATP binding pocket and catalytic residues. Despite lacking kinase activity, Tribbles proteins have a scaffold-like function and participate in protein complex assembly. Through interacting with different kinds of proteins, TRIB3 coordinates crucial cellular processes, including glucose and lipid metabolism, apoptosis, adipocyte differentiation, and cell stress. Our previous study revealed that TRIB3 plays a crucial role in TGFB1-mediated cancer invasion and migration by interacting with the signaling molecule SMAD3. Also, a positive correlation between TRIB3 expression and poor overall survival was reported in breast and colorectal cancer, suggesting that TRIB3 plays a critical role in tumorigenesis and tumor progression.
We have recently demonstrated a positive correlation between TRIB3 and phosphorylated (p-)IRS1 levels in several tumor tissues. Furthermore, the expression levels of TRIB3 and p-IRS1 are negatively correlated with survival rates of patients with these cancers. Metabolic factors, insulin, and IGF1 can induce upregulation of TRIB3, which is responsible for the enhanced tumor proliferation and metastasis in diabetic mice. Overexpression of TRIB3 in normal human bronchial epithelium cells inhibits LC3-I/-II conversion and increases the level of both soluble and insoluble SQSTM1. In contrast, silencing TRIB3 decreases the basal and IGF1-induced accumulation of soluble and insoluble SQSTM1 in HepG2 cells, a human liver carcinoma cell line. The enhanced TRIB3 induces the blockage of autophagic flux leading to an attenuation of the autophagy-lysosome dependent degradation pathway. Hence, accumulation of SQSTM1 and critical tumor-promoting factors promotes tumorigenesis and tumor progression. Interrupting the TRIB3-SQSTM1 interaction by an α-helical peptide derived from SQSTM1 produces an autophagy-inducing effect and potent anti-tumor efficacies ().
Figure 1. Model of the TRIB3-mediated cross-inhibition of autophagy and the UPS in the regulation of metabolic stress-induced tumor development and progression. Metabolic stresses induce upregulation of TRIB3, which interacts physically with SQSTM1 competing for the binding of SQSTM1 to ubiquitinated substrates and LC3. This effect induces the blockage of autophagic flux leading to the accumulation of SQSTM1, which binds ubiquitinated proteins and hinders their proteasomal proteolysis. Both defective autophagy and impaired UPS function act as enhancers of tumor development and progression due to the accumulation of SQSTM1 and cancer-promoting factors. Moreover, the accumulation of EGFR and SQSTM1 reciprocally inhibit autophagic signaling, enhancing the autophagy inhibition effect further. Targeting the TRIB3-SQSTM1 interaction by an α-helical peptide (Pep2-A2) can produce potent antitumor efficacies, suggesting it is a potential strategy against cancers in patients with diabetes.
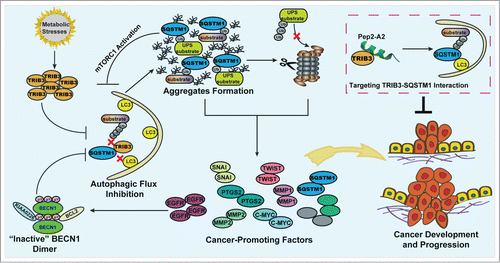
With regard to cancer, autophagy may be either protective, or contribute to progression. Our study indicates that effective autophagic flux is essential for tumor inhibition. Insulin and IGF1, whether or not they activate autophagic signals, induce a high expression level of TRIB3 and a significant accumulation of SQSTM1, one of the critical indicators of a block in autophagic flux, in human liver, colon and lung cancer cells. KK-Ay diabetic mice with high TRIB3 expression also show accumulation of SQSTM1. TRIB3 depletion induces activation of autophagic flux and inhibits tumor metastasis and growth. Furthermore, TRIB3-depleted KK-Ay mice also show decreased SQSTM1 levels and reduced tumor progression. These findings suggest that metabolic stress-enhanced TRIB3 causes autophagic flux attenuation and tumor promotion.
The next question is how TRIB3 inhibits autophagic flux. SQSTM1 is a receptor for delivering ubiquitinated proteins for autolysosomal degradation. As a multidomain protein, SQSTM1 is capable of interacting with several other proteins. Crucial interactions relevant to autophagy are mediated by the C-terminal UBA (ubiquitin interacting) domain and the LIR motif. SQSTM1 and bound ubiquitinated cargos are sequestered inside autophagosomes through the LIR-LC3 interaction. TRIB3 interacts with the LIR-UBA domains of SQSTM1; such interactions are a previously unknown means of regulating LIR-UBA domain interaction partnerships by disrupting the interaction of SQSTM1 with ubiquitinated substrates and LC3. Depletion of TRIB3 relieves SQSTM1 from functional inhibition and prevents the blockage of autophagic flux. In fact, silencing of TRIB3 can induce not only activation of autophagic flux, but also a moderate activation of autophagic signaling. TRIB3-induced inhibition of autophagy results in the accumulation of EGFR and SQSTM1, which reciprocally inhibit autophagic signaling through EGFR-mediated BECN1 phosphorylation and SQSTM1-mediated MTORC1 activation. Interestingly, in our study, the turnover assays revealed that the decrease in tumor-promoting factors resulting from TRIB3 depletion can be reversed in part by bafilomycin A1, by MG132, or by both inhibitors, suggesting that TRIB3 interferes with both autophagic and proteasomal substrate clearance. How can TRIB3 inhibit ubiquitin-proteasome system (UPS) activity? Autophagy defect-induced accumulated SQSTM1 not only binds to ubiquitinated proteins but also competes for binding to these proteins with VCP, which functions to facilitate the delivery of ubiquitinated substrates to the proteasome. Thus, TRIB3-induced autophagy inhibition elevates the accumulation of SQSTM1, which denies the access of ubiquitinated proteins to the proteasome and slows down UPS-specific substrate clearance.
Because the detrimental effects of TRIB3 are predominantly caused by SQSTM1 accumulation and dysfunction, can silencing SQSTM1 recapitulate the effects of TRIB3 depletion? Indeed, silencing TRIB3 inhibits tumor progression not only through decreasing the accumulation of SQSTM1, but more importantly, through interrupting the TRIB3-SQSTM1 interaction and restoring SQSTM1 functions in cancer cells. On the one hand, silencing SQSTM1 decreases the SQSTM1 accumulation, which may inhibit tumor progression; but on the other hand, depletion of SQSTM1 may result in the accumulation of other tumor-promoting factors (such as MMP1, MT-MMP, and TWIST in our current study) due to the abrogation of its autophagic cargo functions. Because of its double-edged sword effect, the comprehensive result of SQSTM1 depletion cannot recapitulate the effects of TRIB3 depletion. Targeting protein-protein interaction is emerging as a promising anticancer strategy. Here, our study provides a candidate α-helical peptide, which disturbs the interaction between TRIB3 and SQSTM1 displaying effective autophagic flux activation and significant antitumor effects. Hence, targeting the interaction between TRIB3 and SQSTM1 is a potential novel strategy against certain cancers.