
Abstract
Autophagy is an important homeostatic process that functions by eliminating defective organelles and aggregated proteins over a neuron's lifetime. One pathological hallmark in amyotrophic lateral sclerosis (ALS)-linked motor neurons (MNs) is axonal accumulation of autophagic vacuoles (AVs), thus raising a fundamental question as to whether reduced autophagic clearance due to an impaired lysosomal system contributes to autophagic stress and axonal degeneration. We recently revealed progressive lysosomal deficits in spinal MNs beginning at early asymptomatic stages in fALS-linked mice expressing the human (Hs) SOD1G93A protein. Such deficits impair the degradation of AVs engulfing damaged mitochondria from distal axons. These early pathological changes are attributable to mutant HsSOD1, which interferes with dynein-driven endolysosomal trafficking. Elucidation of this pathological mechanism is broadly relevant, because autophagy-lysosomal deficits are associated with several major neurodegenerative diseases. Therefore, enhancing autophagic clearance by rescuing endolysosomal trafficking may be a potential therapeutic strategy for ALS and perhaps other neurodegenerative diseases.
Abbreviations
| ALS | = | amyotrophic lateral sclerosis |
| AV | = | autophagic vacuole |
| fALS | = | familial ALS |
| LE | = | late endosome |
| MN | = | motor neuron |
| P40 | = | postnatal day 40 |
| SOD1 | = | superoxide dismutase 1, soluble. |
Amyotrophic lateral sclerosis is one of the most common adult onset MN degenerative disorders. About 10% of ALS cases are familial (fALS), of which over 20% are associated with dominant mutations in genes encoding SOD1 (superoxide dismutase 1, soluble). Mitochondria pathology is a common state that triggers the decline of MN function and survival in ALS-linked pathogenesis. Proper clearance of damaged mitochondria via the autophagy-mitophagy system serves as an early protective mechanism. Autophagic flux is the equilibrium balance between formation and clearance of AVs. Since neurons are particularly sensitive to the accumulation of misfolded proteins and damaged organelles, newly formed AVs are eliminated quickly by fusing with endolysosomes. However, AVs accumulate rapidly in neurons when lysosomal proteolysis is inhibited. Impaired autophagic clearance was reported in various lysosomal storage disorders. Thus, an imbalanced autophagic flux results in autophagic stress characterized by accumulated AVs engulfing protein aggregates and damaged mitochondria. Although increased numbers of AVs are observed in the spinal cords of ALS patients and mutant HsSOD1G93A mice, the contribution of altered autophagy to the ALS-linked pathogenesis has been a subject for debate. It remains unclear (1) whether the observed AVs in fALS-linked MNs reflect enhanced autophagic induction or impaired clearance, and (2) whether enhanced autophagy helps removal of toxic mutant protein aggregates or instead facilitates autophagy stress and MN degeneration. Addressing these issues is critical for future therapeutic strategies for ALS clinical trials.
Lysosomal maturation in neurons depends on the proper retrograde transport of late endosomes (LEs) and endolysosomal trafficking; such dynamic events are particularly challenging in MNs with extended long axons. These raise a fundamental question: does impaired autophagic degradation play an early pathological role during the asymptomatic stage of fALS-linked mice? Our hypothesis is that an imbalanced flux between autophagy induction and subsequent degradation within lysosomes will result in autophagic stress, thus causing distal axons to become more vulnerable to dying-back degeneration. Thus, it is very significant to investigate endolysosomal trafficking and autophagy-lysosomal degradation capacity in the context of both in vivo murine models and in vitro MNs isolated from adult disease-stage mice.
First, we revealed progressive lysosome deficits in HsSOD1G93A spinal MNs with disease progression. There are 4 notable features in our observations: (1) lysosomal deficits are MN-targeted; (2) lysosomal deficits can be detected as early as postnatal day 40 (P40) in asymptomatic HsSOD1G93A mice, and become progressively worse after disease onset at P120; (3) these deficits are not observed in WT HsSOD1 transgenic mice; and (4) the extent of lysosome deficits correlates with levels of AV accumulation. These restricted and progressive changes highlight a pathological link between autophagy-lysosome deficits and MN degeneration.
Second, we examined ultrastructures in spinal cords. Mitochondria in ventral root axons display robust degenerative phenotypes including fragmentation, cristae distortions, vacuolization, or swelling and clustering. These phenotypes are readily detectable as early as in P40 HsSOD1G93A mice. In addition, we observed aberrant clustering of multilamellar bodies and amphisome-like structures engulfing damaged mitochondria with collapsed cristae. By crossing the GFP-LC3 transgenic mouse with the HsSOD1G93A mouse, we further showed colocalization of mitochondria and GFP-LC3-labeled AVs within axon exit zones, suggesting impaired autophagy-mitophagy degradation in MN axons of HsSOD1G93A mice at early asymptomatic stages.
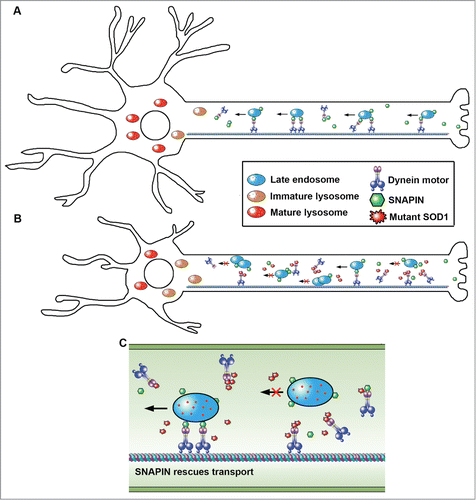
We next addressed whether lysosome deficits impair autophagy clearance by characterizing lysosomal maturation and autophagy degradation in live spinal MN cultures isolated from adult diseased mice. Compared to embryonic MN cultures, adult MNs provide more reliable cell models for investigating cellular mechanisms underlying adult onset pathogenesis. Consistently, cultured adult HsSOD1G93A MNs recapitulate lysosome deficits and impaired AV degradation. We took advantage of live spinal MN cultures to examine axonal transport of late endocytic organelles. HsSOD1G93A MNs display reduced retrograde transport. Such early transport deficits are due to competitive binding of mutant HsSOD1G93A to DNAI (dynein, axomenal, intermediate chain), and can be rescued by introducing a Snapin transgene in MNs. SNAPIN acts as an adaptor recruiting dynein motors to LEs via binding to DNAI, thus coordinating dynein-driven retrograde transport and endolysosomal trafficking. Overexpressing SNAPIN in HsSOD1G93A MNs enhances LE retrograde transport, thus reducing their clustering in axons. SNAPIN competes with HsSOD1G93A for binding to dynein DNAI, thereby recruiting dynein motors to LEs for retrograde transport. Thus, SNAPIN and HsSOD1G93A play opposite roles in LE retrograde transport.
Intriguingly, early deficits in autophagy-lysosomal function in HsSOD1G93A MNs can be effectively rescued by SNAPIN expression both in vitro and in vivo. SNAPIN competes with HsSOD1G93A for binding to dynein motors and rescues endolysosomal trafficking and lysosome maturation, thereby removing damaged mitochondria from axons and prolonging MN survival. Injecting adeno-associated virus serotype 9/AAV9-SNAPIN into the diseased mouse spinal cords rescues lysosome deficits in MNs in vivo and slows MN degeneration and disease progression. Thus, our study advances our understanding of early pathological mechanisms underlying MN degeneration and provides new mechanistic insights into how: (1) mutant HsSOD1 impairs LE retrograde transport by interfering with the motor-cargo coupling; and (2) SNAPIN reverses the mutant phenotypes by competing with HsSOD1G93A for dynein-driven LE retrograde transport ().
Figure 1. Mutant HsSOD1G93A and SNAPIN play opposite roles in the dynein-driven retrograde transport of late endosomes and lysosome maturation. (A) SNAPIN acts as an adaptor recruiting dynein motors to late endosomes via binding to DNAI, thus coordinating dynein-driven retrograde transport toward the soma and endolysosomal trafficking and lysosomal maturation. This mechanism enables neurons to maintain efficient autophagy-lysosomal degradation capacity. (B) In mutant HsSOD1G93A motor neurons, the SNAPIN-dynein coupling is blocked by HsSODG93A-DNAI interaction, thereby reducing dynein recruitment to late endosomes for driving retrograde transport toward the soma, and impairing lysosome maturation. These phenotypes are readily detectable at early asymptomatic stages of HsSOD1G93A mice. (C) Elevated SNAPIN expression reverses the mutant phenotype by competing with mutant HsSOD1G93A for binding dynein DNAI. By competitively binding to dynein, mutant HsSOD1G93A and SNAPIN play opposite roles in the dynein-driven retrograde transport of late endosomes, and thus autophagy-lysosome function.
In summary, our study provides in vitro and in vivo evidence that progressive lysosomal deficits combined with impaired autophagic clearance are the early fALS-linked pathological events. Our findings reveal mechanistic clues as why inducing autophagy alone, without compensatory rescue of LE transport and lysosomal deficits, fails to reduce mutant HsSOD1 aggregates. Instead it augments autophagy stress and mitochondria pathology. The dynein-SNAPIN (motor-adaptor) coupling may help remove axonal AVs. Accumulation of AVs engulfing dysfunctional mitochondria within distal axons early in the disease course can have catastrophic consequences by triggering axonal degeneration and denervation. This view is consistent with the notion that ALS is a dying-back type of neuropathy that initiates and progresses from distal to proximal portions of MNs. Therefore, enhancing lysosome function, rather than autophagy induction, is an alternative and promising therapeutic strategy for ALS-linked clinical trials.