
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Loss-of-function mutations in the gene encoding GBA (glucocerebrosidase, β, acid), the enzyme deficient in the lysosomal storage disorder Gaucher disease, elevate the risk of Parkinson disease (PD), which is characterized by the misprocessing of SNCA/α-synuclein. However, the mechanistic link between GBA deficiency and SNCA accumulation remains poorly understood. In this study, we found that loss of GBA function resulted in increased levels of SNCA via inhibition of the autophagic pathway in SK-N-SH neuroblastoma cells, primary rat cortical neurons, or the rat striatum. Furthermore, expression of the autophagy pathway component BECN1 was downregulated as a result of the GBA knockdown-induced decrease in glucocerebrosidase activity. Most importantly, inhibition of autophagy by loss of GBA function was associated with PPP2A (protein phosphatase 2A) inactivation via Tyr307 phosphorylation. C2-ceramide (C2), a PPP2A agonist, activated autophagy in GBA-silenced cells, while GBA knockdown-induced SNCA accumulation was reversed by C2 or rapamycin (an autophagy inducer), suggesting that PPP2A plays an important role in the GBA knockdown-mediated inhibition of autophagy. These findings demonstrate that loss of GBA function may contribute to SNCA accumulation through inhibition of autophagy via PPP2A inactivation, thereby providing a mechanistic basis for the increased PD risk associated with GBA deficiency.
Abbreviations
| ACTB | = | actin, β |
| AD | = | Alzheimer disease |
| ATG5 | = | autophagy-related 5 |
| BafA1 | = | bafilomycin A1 |
| BECN1 | = | Beclin 1, autophagy related |
| C2 | = | C2-ceramide |
| GBA | = | glucosidase, β, acid |
| GD | = | Gaucher disease |
| GlcCer | = | glucosylceramide |
| LBD | = | Lewy body disorders |
| LBs | = | Lewy bodies |
| LV | = | lentivirus |
| MAP1LC3A/LC3 | = | microtubule-associated protein 1 light chain 3 |
| OA | = | okadaic acid |
| PD | = | Parkinson disease |
| PPP2A | = | protein phosphatase 2A |
| PPP2C | = | protein phosphatase 2, catalytic subunits |
| PPP2R | = | protein phosphatase 2, regulatory subunits |
| PPP2R1 | = | protein phosphatase 2, regulatory subunit A |
| Rapa | = | rapamycin |
| SNCA | = | synuclein, α (non A4 component of amyloid precursor) |
| TEM | = | transmission electron microscopy. |
Introduction
Parkinson disease (PD) is the second most common progressive neurodegenerative disorder, and is characterized by the loss of dopaminergic neurons in the substantia nigra pars compacta and the presence of Lewy bodies (LBs) in vulnerable populations of neurons.Citation1 The major protein component of LBs is SNCA, a synaptic protein with the propensity to misfold and aggregate. Three missense mutations (A53T, A30P, and E46K) in the gene encoding SNCA cause an autosomal dominant form of PD.Citation2 Numerous susceptibility genes have been shown to confer predisposition to PD, including SNCA, PARK2, PINK1, PARK7, and LRRK2.Citation3 In recent years, mutations in the gene encoding GBA (glucosidase, β, acid) have been identified as the most common known genetic risk factor for the development of PD.Citation4-7
Gaucher disease (GD) is a rare, autosomal recessive lysosomal storage disorder that results from loss-of-function mutations in GBA. GBA is a lysosomal enzyme responsible for the conversion of glucocerebroside to glucose and ceramide.Citation8 The human GBA gene maps to 1q21, and consists of 11 exons encoding a 497 amino acid protein. Nearly 300 mutations have been identified in GD patients.Citation9 Accumulating evidence over the past decade has revealed an association between mutations in GBA and the development of PD and other Lewy body disorders (LBD). Clinical studies have repor3ted parkinsonism among relatives of patients with GD,Citation10 while genotyping studies have demonstrated a higher incidence of GBA mutations in cohorts of PD patients of different ethnic origins, particularly those with early onset PD.Citation7,11-13 These findings suggest a possible mechanistic link between PD and GD. Recently, neuropathological analysis of GD patients revealed the presence of SNCA-positive LBs,Citation14 suggesting that GBA may contribute to the aggregation of SNCA. Postmortem examination of PD brains with GBA mutations revealed reduced levels of GBA protein, primarily in the substantia nigra.Citation15 GBA knockdown in neurons led to decreased lysosomal degradation capacity, and increased levels of SNCA protein.Citation16 However, the mechanism underlying the accumulation of SNCA as a result of GBA deficiency is still unclear.
A recent in vitro study has found that GBA mutations lead to SNCA accumulation, which can be pharmacologically reversed by rapamycin, an inducer of macroautophagy.Citation17 In addition, macroautophagy also participates in SNCA degradation.Citation18 These findings suggest that loss of GBA function could promote SNCA accumulation through inhibition of the autophagic pathway.
PPP2A (protein phosphatase 2A) exists as a heterotrimer consisting of a 36-kDa catalytic subunit C, PPP2CA or PPP2CB (referred to here as PPP2C), a 65-kDa structural subunit A (a scaffolding subunit), PPP2R1A or PPP2R1B, and a variable regulatory subunit B, PPP2R (with various isoforms). Modifications in PPP2C are important for the regulation of phosphatase activity: phosphorylation of PPP2C at Tyr307 inhibits PPP2A activity by 90% in vitro.Citation19 PPP2A is an important regulator of numerous cellular processes, including cell growth and proliferation,Citation20 apoptosis,Citation21 transcription, and translation.Citation22 Recent studies indicate that PPP2A is involved in the regulation of rapamycin-induced autophagy in yeast,Citation23 although the precise role of PPP2A in GBA deficiency remains to be elucidated.
In this study, the mechanistic basis of SNCA accumulation induced by loss of GBA function was examined in vitro (in SK-N-SH neuroblastoma cells or primary rat cortical neurons) and in vivo (in the rat striatum). The results provide the first evidence that GBA deficiency inhibits the autophagic pathway by decreasing PPP2A activity and promotes SNCA accumulation, thereby demonstrating a link between loss of GBA function and increased PD risk.
Results
Loss of GBA function leads to SNCA accumulation in neuroblastoma cells
A previous study has shown that loss-of-function mutations in GBA promote SNCA accumulation in vitro.Citation17 To analyze the effect of GBA deficiency on SNCA levels, GBA expression was suppressed in SK-N-SH neuroblastoma cells using plasmids expressing shRNA against human GBA. The efficiency of knockdown for 2 different GBA shRNA pairs (shGBA #9847–1 and shGBA #9848–1) was measured at 12, 24, and 48 h (). Expression of the~60 kDa GBA protein was lower at 12 and 24 h compared to control cells upon transfection of shGBA #9847–1 and shGBA #9848–1, although a recovery of normal protein expression was observed at 48 h. At 12 h, GBA expression was reduced to 40% (shGBA #9847–1) and 36% (shGBA #9848–1) of the level in control cells, and was further reduced to 34% (shGBA #9847–1) and 29% (shGBA #9848–1) at 24 h (). Based on these results, shGBA #9847–1 and shGBA #9848–1 were used to transfect cells for 24h in subsequent experiments.
Figure 1. ShRNA-mediated knockdown of GBA leads to SNCA accumulation in SK-N-SH neuroblastoma cells. (A) Efficiency of GBA knockdown with shGBA #9847–1 and shGBA #9848–1 was assessed at 12, 24, and 48 h. (B) GBA protein expression in cells transfected with shRNA constructs was reduced at 12 and 24 h. (C) GBA activity was reduced in SK-N-SH neuroblastoma cells transfected with shGBA #9847–1 and shGBA #9848–1. *P< 0.05 vs. normal; n = 3. (D, E) Increase in SNCA protein expression in GBA knockdown cells. (F, G) GBA and SNCA mRNA expression after GBA knockdown was assessed by quantitative RT-PCR. *P< 0.05, **P< 0.01 vs. normal; n = 3.
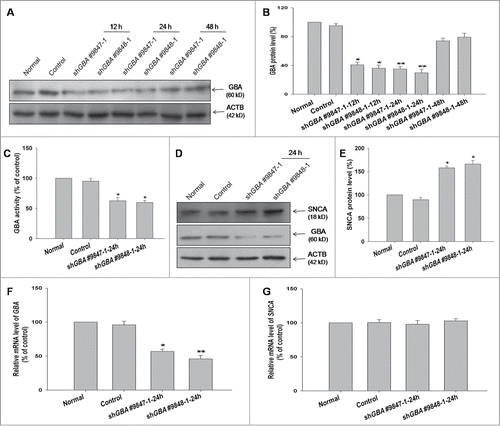
Since LBs have been detected in postmortem brain samples of GD patients, the accumulation of SNCA in SK-N-SH cells transiently transfected with GBA shRNA was investigated. As expected, GBA activity in SK-N-SH cell lysates decreased to approximately 60% of the control level upon GBA knockdown using both shGBA #9847–1 and shGBA #9848–1 (). GBA knockdown was accompanied by a 1.6fold increase in SNCA protein level relative to the control (). To determine whether increased levels of SNCA protein were due to transcriptional upregulation, SNCA mRNA levels in GBA-silenced SK-N-SH cells were measured by RT-PCR. No changes in SNCA mRNA level were detected, suggesting that the observed increase in SNCA protein level resulted from compromised protein degradation (). Therefore, the role of the autophagic pathway on GBA knockdown-induced SNCA accumulation was examined in greater detail.
GBA knockdown inhibits autophagy in neuroblastoma cells and cortical neurons
In autophagosome formation, the cytosolic form of microtubule-associated protein 1 light chain 3/LC3-I is converted to a lipid-conjugated LC3-II form; the amount of LC3-II correlates with autophagosome number and is therefore considered as a simple and reliable measure of autophagic activity in mammalian cells.Citation24 The level of LC3-II was assessed in GBA-silenced SK-N-SH cells by western blotting at 24 h. Knockdown of GBA using both shGBA #9847–1 and shGBA #9848–1 resulted in decreased LC3-II compared to control cells (). These results indicate that loss of GBA inhibits autophagy in SK-N-SH cells.
Figure 2. GBA knockdown inhibits autophagy in SK-N-SH neuroblastoma cells and rat cortical neurons. (A) Decreased LC3-II level in cells transfected with shGBA #9847–1 or shGBA #9848–1. (B) GBA protein expression was reduced in GBA knockdown cells. (C) Decrease in autophagic activity upon GBA knockdown, as assessed by the ratio of LC3-II/ACTB. **P< 0.01 compared to normal; n = 3. (D, E) Efficiency of GBA knockdown upon infection of neurons with different LV. LV-GFP-shGba #6 showed the greatest effect, causing a 50% reduction in GBA protein level. (F) GBA activity was reduced in cortical neurons infected with LV-GFP-shGba #6, an effect that was reversed by coinfection with full-length GBA (LV-GFP-shGba #6+HsGBA). *P< 0.05 vs. LV-GFP-sh-con group, #P< 0.05 vs. LV-GFP-shGba #6 group; n = 3. (G) Decreased LC3-II level upon GBA knockdown. Neurons treated with rapamycin (LV-GFP-shGba #6+Rapa; 40 nM for 6 h) served as a positive control. (H) GBA protein expression was reduced in cells infected with LV-GFP-shGba #6, an effect that was rescued by coinfection of full-length GBA (LV-GFP-shGba #6+HsGBA). (I) Decrease in autophagic activity upon GBA knockdown in LV-GFP-shGba #6-infected neurons, as assessed by the ratio of LC3-II/ACTB. Activity was restored to a normal level upon overexpression of full-length GBA (LV-GFP-shGba #6+HsGBA) or rapamycin treatment (LV-GFP-shGba #6+Rapa). *P< 0.05, **P< 0.01 compared to LV-GFP-sh-con group, #P< 0.05, ###P< 0.001 compared to LV-GFP-shGba #6 group; n = 3.
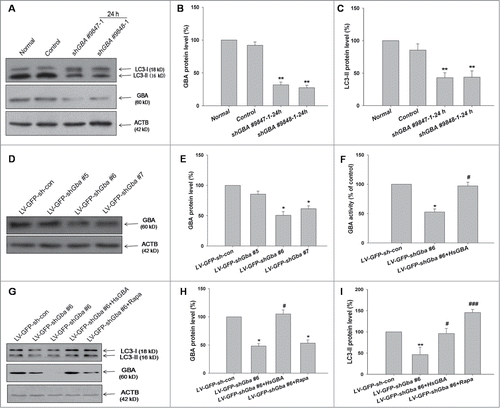
The effect of GBA knockdown on autophagic activity was examined in primary rat cortical neurons. Neurons were infected with lentivirus (LVs) encoding GFP–shRNAs (LV-GFP-shGba #5, LV-GFP-shGba #6, and LV-GFP-shGba #7) and scrambled negative control (LV-GFP-sh-con). The efficiency of GBA knockdown was assessed 3 d after infection. The level of GBA protein was reduced by 50% and 39% in neurons infected with LV-GFP-shGba #6 and LV-GFP-shGba #7, respectively, compared to control neurons (), while only a modest reduction (14%) was observed using LV-GFP-shGba #5. Thus, LV-GFP-shGba #6 was used in subsequent experiments. As predicted, a reduction in GBA activity (47%) was also observed in LV-GFP-shGba #6-infected cortical neurons, which was rescued by coinfection with LV-GFP-HsGBA (). These data confirmed that GBA activity was inhibited by GBA knockdown. GFP fluorescence was robust 3 d after infection, indicating efficient infection and low toxicity (Fig. S1).
The level of LC3-II was detected by western blotting (). GBA protein expression was reduced in neurons infected with LV-GFP-shGba #6, but was rescued by coinfection with full-length GBA (LV-GFP-shGba #6+HsGBA; ). Autophagic activity, as assessed by the level of LC3-II, was lower in LV-GFP-shGba #6-infected neurons; this effect was rescued by overexpression of full-length GBA and treatment with rapamycin, an autophagy agonist (40 nM for 6 h) (). These results indicate that loss of GBA function inhibits autophagy in cultured cortical neurons, consistent with the effects observed in SK-N-SH cells.
GBA knockdown decreases autophagosome formation
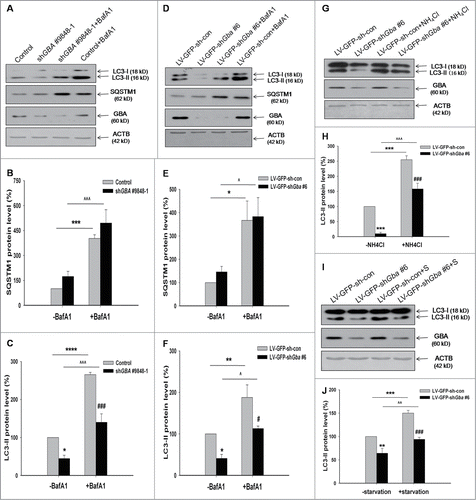
LC3-II was known as a marker of autophagosome. Decreased LC3-II could indicate either decreased formation of autophagosomes or increased flux of autophagosomes to autolysosomes. Efficient flux of autophagy would result in a decrease in the substrates, such as SQSTM1/P62, a polyubiquitin-binding protein. On the contrary, SQSTM1 showed a slight increase in GBA-deficient SK-N-SH cells and neurons compared to the control group, but the change was not statistically significant (), suggesting the decreased formation of autophagosomes. To further evaluate the autophagic flux through the macroautophagic pathway, cells were treated with 100 nM bafilomycin A1 (BafA1) for 6 h (). BafA1 inhibits vacuolar-type ATPases and impairs vesicle fusion; the final steps of autophagy are thereby obstructed due to the failure of autophagosome-lysosome fusion.Citation25 In the presence of BafA1, a decrease in LC3-II level and no obvious changes in SQSTM1 level were observed in GBA-deficient SK-N-SH cells and neurons compared to the control group (), indicating that the decrease in LC3-II upon GBA knockdown was due to decreased formation of autophagosomes. NH4Cl, a well-established lysosomal inhibitor, also reduced LC3-II level in GBA-deficient neurons, as compared to the control group treated with NH4Cl (10 mM for 1 h) (). In addition, the LC3-II level was analyzed in response to starvation, a more physiologically relevant treatment. Similarly, the LC3-II level was decreased in response to starvation in GBA-deficient neurons ().
Figure 3. Assessment of autophagic flux, following GBA knockdown. Autophagic flux was analyzed in GBA-deficient SK-N-SH neuroblastoma cells (A, B, C) and cortical neurons (D, E, F) treated with 100 nM BafA1 for 6 h or 10 mM NH4Cl for 1 h (G, H). The levels of LC3-II and SQSTM1 were normalized with ACTB. *P< 0.05, ***P< 0.001, ****P< 0.0001 vs. control; ###P< 0.001 vs. control+BafA1 group; *P< 0.05, **P< 0.01, ***P< 0.001 vs. LV-GFP-sh-con group; #P< 0.05 vs. LV-GFP-sh-con+BafA1 group; ###P< 0.001 vs. LV-GFP-sh-con+ NH4Cl group; ^P< 0.05, ^^^P< 0.001 vs. LV-GFP-shGba #6 group; ^^^P< 0.001 vs. shGBA #9848–1 group; n = 3. (I, J) LC3-II level was analyzed in response to starvation 2 h (Hank's balanced salt solution). ACTB was used as a loading control. **P< 0.01, ***P< 0.001 vs. LV-GFP-sh-con group; ###P< 0.001 vs. LV-GFP-sh-con+starvation group; ^^P< 0.01 vs. LV-GFP-shGba #6 group; n = 3.
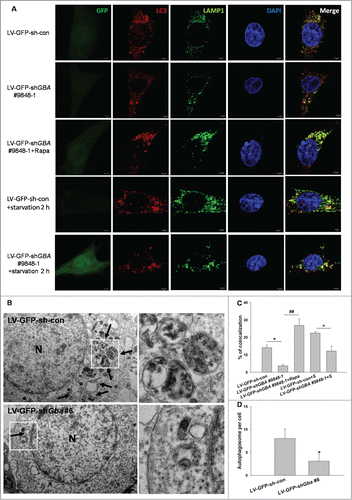
We next measured the colocalization of LC3 and LAMP1 by using an immunocytochemical method in SK-N-SH neuroblastoma cells. A decrease in the colocalization of LC3 and LAMP1 was detected in GBA-deficient SK-N-SH cells (). Furthermore, electron microscopy (EM) analysis verified a decrease in the number of autophagosomes in GBA-deficient neurons (). Taken together, these results suggested that loss of GBA function inhibited autophagy through decreasing the formation of autophagosomes.
Figure 4. GBA knockdown inhibits autophagosome formation. (A) SK-N-SH neuroblastoma cells were infected with LV-GFP-shGBA #9848–1. After 3 d, cells were fixed and stained with LAMP1 and LC3 primary antibodies. Immunolabeling was visualized with rabbit Alexa 647-conjugated and mouse Alexa Fluor 594-conjugated secondary antibodies, respectively. The nucleus was counterstained by DAPI. These detected cells were first verified to infect GFP. Then the channel was turned off. To better observe the colocalization of LAMP1 and LC3, LAMP1 was labeled as “green.” Scale bar =2 μm. (B) TEM images of autophagic vacuoles in primary rat cortical neurons. The cortical neurons were infected with LV-GFP-shGba #6 for 3 d The right panel is a high magnification image of the indicated portion. Arrows indicate autophagosomes. N, nuclei. Scale bar =500 nm. (C) Fluorescence images were analyzed using ImageJ “colocalization” plug-in software. A total of 30 cells were analyzed from 3 independent experiments (10 cells per experiment). The colocalization of LAMP1 and LC3 was decreased in GBA-deficient cells compared to control group, which was reversed by treatment with rapamycin (40 nM for 6 h). Also, the colocalization of LAMP1 and LC3 was decreased in GBA-deficient cells in response to starvation. *P< 0.05, ##P< 0.01, ^P< 0.05. (D) TEM revealed less autophagosomes in neurons infected with LV-GFP-shGba #6 than in LV-GFP-sh-con group. Twenty cells per experiment, n = 3. *P< 0.05.
Expression of the autophagy pathway component BECN1 is downregulated by GBA knockdown
As shown above, GBA knockdown inhibited its activity; this leads to the accumulation of the substrates glucosylceramide (GlcCer) and glucosylsphingosine, and a diminished production of ceramide.Citation8 Intracellular ceramide and GlcCer level was assessed in GBA-deficient SK-N-SH cells by immunocytochemistry and by measuring C2 concentration in the lipid fraction by HPLC-MS/MS. GBA knockdown using LV-sh-9848–1# resulted in a decreased ceramide level and an increase GlcCer level relative to control cells ( and S2). In lipid extracts, endogenous C2 level was reduced in GBA-deficient cells, an effect that was reversed upon application of exogenous C2 ().These results indicate that ceramide production is reduced in the absence of GBA.
Figure 5. Reduction in ceramide production and downregulation of BECN1 protein expression upon GBA knockdown. (A) SK-N-SH neuroblastoma cells were infected with LV-GFP-shGBA #9848–1 for 3 d, and treated with an antibody against ceramide and stained with DAPI. Scale bar =50 μm. (B) Reduced ceramide production in cells infected with LV-GFP-shGBA #9848–1. Samples were imaged at the same exposure to allow direct comparisons; ceramide staining intensity was calculated using ImageJ software. Data were analyzed for 10 randomly selected fields of view from 3 independent experiments. **P< 0.01 vs. LV-GFP-sh-con group. (C) Endogenous C2 level was reduced in GBA-deficient SK-N-SH neuroblastoma cells infected with LV-GFP-shGBA #9848–1, which was reversed by exogenous C2 application (5 μM for 8 h), as determined by HPLC-MS/MS. **P< 0.01 vs. LV-GFP-sh-con group, ##P< 0.01 vs. LV-GFP-shGBA #9848–1 (n=3). (D, E) Downregulation of BECN1 protein expression in GBA knockdown neuroblastoma cells transfected with shGBA #9847–1 and shGBA #9848–1. *P< 0.05, **P< 0.01 vs. normal; n = 3. (F, G) Decreased expression of BECN1 protein induced by GBA knockdown in cortical neurons is rescued by overexpression of full-length GBA (LV-GFP-shGba #6+HsGBA) or treatment with rapamycin (LV-GFP-shGba #6+Rapa; 40 nM for 6 h). **P< 0.01 vs. LV-GFP-sh-con group, #P< 0.05, ##P< 0.01 vs. LV-GFP-shGba #6 group; n = 3.
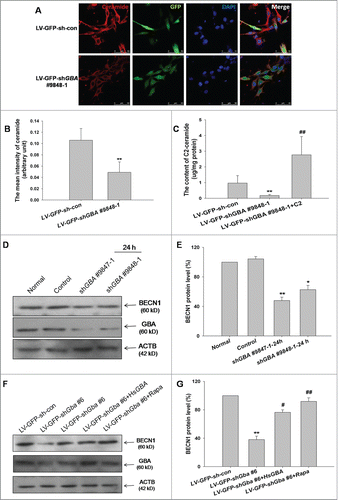
Ceramide stimulates autophagy by inducing the expression of BECN1.Citation26 Therefore, the effect of GBA knockdown on BECN1 protein level was examined. BECN1 expression was reduced relative to the control 24 h after transfection of SK-N-SH cells with shGBA #9847–1 and shGBA #9848–1 (). Similarly, BECN1 expression was decreased in cortical neurons infected with LV-GFP-shGba #6, an effect that was reversed by the overexpression of full-length GBA or treatment of neurons with rapamycin (). BECN1 protein level was further assessed in GBA-deficient neurons treated with exogenous C2. GBA knockdown resulted in a decreased BECN1 expression relative to control neurons, which was reversed by application of exogenous C2 (Fig. S3). Taken together, these results suggest that expression of the autophagy pathway component BECN1 is downregulated as a result of the GBA knockdown-induced decrease in GBA activity.
Inhibition of autophagy by loss of GBA involves PPP2A inactivation via Tyr307 phosphorylation
Results from previous studies suggest that downregulation of PPP2A, perturbation of autophagy, and the presence of ubiquitinated protein inclusions are hallmarks of neurodegenerative diseases such as Alzheimer disease (AD).Citation27,28 However, the effect of GBA deficiency on PPP2A activity has never been examined. PPP2A activity was therefore measured in SK-N-SH cells and cortical neurons following knockdown of GBA. PPP2A activity decreased in neuroblastoma cells transfected with shGBA #9847–1 or shGBA #9848–1, and neurons infected with LV-GFP-shGba #6 (), which was reversed by simultaneously expressing full-length GBA.
Figure 6. Inactivation of PPP2A via Tyr307 phosphorylation induced by loss of GBA function. (A) PPP2A activity was reduced in SK-N-SH neuroblastoma cells transfected with shGBA #9847–1 and shGBA #9848–1. **P< 0.01 compared to normal; n = 3. (B) PPP2A activity was reduced in cortical neurons infected with LV-GFP-shGba #6, which was reversed by coinfection of cells with full-length GBA (LV-GFP-shGba #6+HsGBA) or C2 treatment (LV-GFP-shGba #6+C2; 5 μM for 8 h). **P< 0.01 compared to LV-GFP-sh-con group, ##P< 0.01 compared to LV-GFP-shGba #6 group; n = 3. ((C)to E) Increased phosphorylation of PPP2A at Tyr307 (p-PPP2A [Tyr307]) in neuroblastoma cells transfected with shGBA #9847–1 and shGBA #9848–1. *P< 0.05 compared to normal; n = 3. ((F)to H) Increased level of p-PPP2A (Tyr307) in cortical neurons infected with LV-GFP-shGba #6; the effect was abolished by overexpression of full-length GBA (LV-GFP-shGba #6+HsGBA) or C2 treatment (LV-GFP-shGba #6+C2). **P< 0.01 compared to LV-GFP-sh-con group, ##P< 0.01 compared to LV-GFP-shGba #6 group; n = 3.
![Figure 6. Inactivation of PPP2A via Tyr307 phosphorylation induced by loss of GBA function. (A) PPP2A activity was reduced in SK-N-SH neuroblastoma cells transfected with shGBA #9847–1 and shGBA #9848–1. **P< 0.01 compared to normal; n = 3. (B) PPP2A activity was reduced in cortical neurons infected with LV-GFP-shGba #6, which was reversed by coinfection of cells with full-length GBA (LV-GFP-shGba #6+HsGBA) or C2 treatment (LV-GFP-shGba #6+C2; 5 μM for 8 h). **P< 0.01 compared to LV-GFP-sh-con group, ##P< 0.01 compared to LV-GFP-shGba #6 group; n = 3. ((C)to E) Increased phosphorylation of PPP2A at Tyr307 (p-PPP2A [Tyr307]) in neuroblastoma cells transfected with shGBA #9847–1 and shGBA #9848–1. *P< 0.05 compared to normal; n = 3. ((F)to H) Increased level of p-PPP2A (Tyr307) in cortical neurons infected with LV-GFP-shGba #6; the effect was abolished by overexpression of full-length GBA (LV-GFP-shGba #6+HsGBA) or C2 treatment (LV-GFP-shGba #6+C2). **P< 0.01 compared to LV-GFP-sh-con group, ##P< 0.01 compared to LV-GFP-shGba #6 group; n = 3.](/cms/asset/6d2f280d-8e42-4574-b387-0ea516e76326/kaup_a_1086055_f0006_b.gif)
To elucidate the mechanism of PPP2A inactivation, the phosphorylation status of PPP2A at Tyr307 (p-PPP2A-Tyr307, representing an inactive form of PPP2A) was examined. In addition, C2, an agonist of PPP2A, was used to stimulate PPP2A activity in GBA-silenced cells. The optimal C2 concentration (5 μM for 8 h) was determined in a previous study (Fig. S4). The phosphorylation of Tyr307 increased in SK-N-SH cells and cortical neurons upon knockdown of GBA (), indicating an inhibition of PPP2A activity. The increase in p-PPP2A-Tyr307 level was abolished in the presence of full-length GBA or C2 ().
To further confirm the contribution of PPP2A activity to autophagic pathway inhibition resulting from GBA deficiency, LC3-II was measured in conjunction with Tyr307 phosphorylation and PPP2C expression (). A decrease in LC3-II, and an increase in Tyr307 phosphorylation, was observed upon knockdown of GBA in SK-N-SH cells relative to controls; however, a normal LC3-II and p-PPP2A-Tyr307 expression level was restored with C2 treatment (). These results indicate that the inhibition of autophagy induced by loss of GBA is through suppression of PPP2A activity via Tyr307 phosphorylation.
Figure 7. C2 treatment restores autophagic activity in SK-N-SH neuroblastoma cells with GBA knockdown. (A to C) Decreased LC3-II level, as well as increased PPP2A phosphorylation at Tyr307 (p-PPP2A [Tyr307]) in cells transfected with shGBA #9848–1 were reversed by C2 treatment (shGBA #9848–1+C2; 5 μM for 8 h). (D) Autophagic activity was restored in GBA-deficient cells in the presence of C2, as assessed by the ratio of LC3-II/ACTB. **P< 0.01, ***P< 0.001 compared to normal, ##P< 0.01 compared to shGBA #9848–1 group; n = 3.
![Figure 7. C2 treatment restores autophagic activity in SK-N-SH neuroblastoma cells with GBA knockdown. (A to C) Decreased LC3-II level, as well as increased PPP2A phosphorylation at Tyr307 (p-PPP2A [Tyr307]) in cells transfected with shGBA #9848–1 were reversed by C2 treatment (shGBA #9848–1+C2; 5 μM for 8 h). (D) Autophagic activity was restored in GBA-deficient cells in the presence of C2, as assessed by the ratio of LC3-II/ACTB. **P< 0.01, ***P< 0.001 compared to normal, ##P< 0.01 compared to shGBA #9848–1 group; n = 3.](/cms/asset/709f4de9-1323-43ee-8214-729d5195c64a/kaup_a_1086055_f0007_b.gif)
Loss of GBA function induces the accumulation of SNCA via inhibition of the autophagy pathway
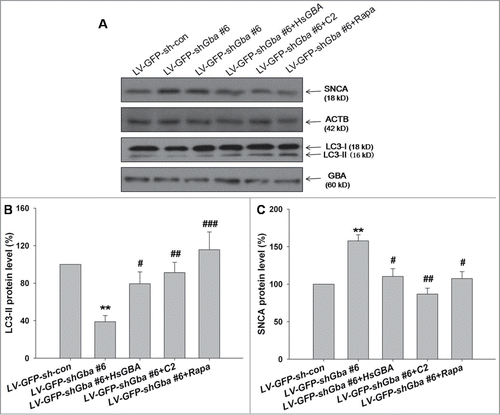
To assess the role of autophagic activity in GBA knockdown-induced SNCA accumulation, expression of SNCA protein was assessed in GBA-deficient cortical neurons in the presence of agonists of PPP2A or autophagy. Protein expression of SNCA increased in LV-GFP-shGba #6-infected neurons; this effect was abolished upon coinfection of full-length GBA, or treatment with C2 or rapamycin (). Autophagic activity, which was suppressed in knockdown cells, was similarly restored to normal levels by application of C2 or rapamycin, which was accompanied by a decline in SNCA protein accumulation (). To further clarify the role of macroautophagy pathway on SNCA accumulation, neurons were infected with LV-cherry-shAtg5 and scrambled negative control (LV-cherry-sh-con). Accordingly, the level of ATG5 protein was reduced in neurons infected with LV-cherry-shAtg5, compared to control neurons. Moreover, Atg5 knockdown led to SNCA accumulation, which was abolished upon treatment with rapamycin (40 nM for 6 h) (Fig. S5). These results suggest the causal relationship between SNCA accumulation and macroautophagy. Taken together, these findings demonstrate that loss of GBA function results in the suppression of autophagy via inhibition of PPP2A activity, thereby leading to SNCA accumulation.
Figure 8. Loss of GBA function leads to inhibition of autophagy via downregulation of PPP2A activity, resulting in SNCA accumulation in cortical neurons. (A, B) Increased SNCA protein expression, and decreased autophagic activity (LC3-II/ACTB), in neurons infected with LV-GFP-shGba #6 are reversed by overexpression of full-length GBA (LV-GFP-shGba #6+HsGBA), or by treatment with C2 (LV-GFP-shGba #6+C2; 5 μM for 8 h) or rapamycin (LV-GFP-shGba #6+Rapa; 40 nM for 6 h). (C) Expression of SNCA protein in GBA-deficient neurons was restored to normal levels by overexpression of full-length GBA, or treatment with PPP2A or autophagy agonists. **P< 0.01 compared to LV-GFP-sh-con group, #P< 0.05, ##P< 0.01, ###P< 0.001 compared to LV-GFP-shGba #6 group; n = 3.
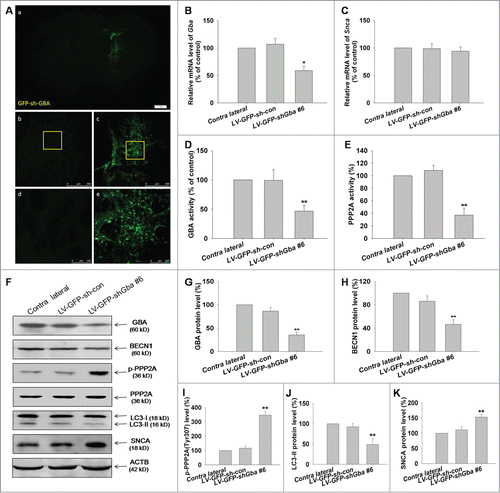
SNCA accumulation induced by loss of GBA function involves inhibition of autophagy via PPP2A inactivation in rats
To confirm the mechanistic basis of SNCA accumulation induced by loss of GBA function in vivo, LV gene transfer vectors encoding GFP–shGba #6 (LV-GFP-shGba #6) and scrambled negative control (LV-GFP-sh-con) were used to infect the left striatum of rats. LV infection of the striatal tissue was confirmed by GFP expression (). The efficiency of GBA knockdown was assessed 1 week after infection; the level of GBA protein was reduced to 35% in the left striatum as compared to the contralateral side. GBA knockdown was accompanied by a 1.5-fold increase in SNCA protein level relative to that of the contralateral striatum (), while no changes in Snca transcript expression were detected by RT-PCR (), suggesting that the observed increase in SNCA protein level resulted from compromised protein degradation, consistent with the in vitro results. Furthermore, GBA and PPP2A activities were markedly reduced in the striatum of rats infected with LV-GFP-shGba #6 (), while the phosphorylation of PPP2A at Tyr307 was increased (). A decrease in BECN1 expression and the LC3-II level was also observed upon knockdown of GBA in the striatum relative to the contralateral side (). Taken together, these results indicate that the loss of GBA function promotes SNCA accumulation through inhibition of the autophagy pathway via PPP2A inactivation.
Figure 9. Loss of GBA function leads to SNCA accumulation through inhibition of autophagy involving PPP2A inactivation in rats. (A) Confocal immunofluorescence microscopy reveals an enhancement of GFP signal following LV-GFP-shGba #6 injection into the left striatum of rats, with no fluorescence detected on the contralateral (uninjected) side. Scale bar = 500 μm. (B, C) LV gene transfer vectors encoding GFP-shGba #6 (LV-GFP-shGba #6) and scrambled negative control (LV-GFP-sh-con) were injected into the left striatum of rats; 1 wk later, RNA was extracted from whole striatal tissue homogenates for analysis of Gba and Snca mRNA expression by quantitative RT-PCR. (D, E) GBA and PPP2A activities were reduced in the striatum of rats infected with LV-GFP-shGba #6. (F) GBA expression, autophagy level, phosphorylation level of PPP2A at Tyr307, and SNCA expression were assessed by western blotting. (G) GBA protein expression was reduced in the striatum of rats infected with LV-GFP-shGba #6. ((H)to K) Downregulation of BECN1 protein expression, increased phosphorylation of PPP2A at Tyr307, decreased LC3-II level, and increased SNCA protein expression were observed in the striatum infected with LV-GFP-shGba #6. *P< 0.05, **P< 0.01 vs. contralateral group; n = 3.
Loss of GBA function leads to the production and aggregation of SNCA oligomeric species in cortical neurons
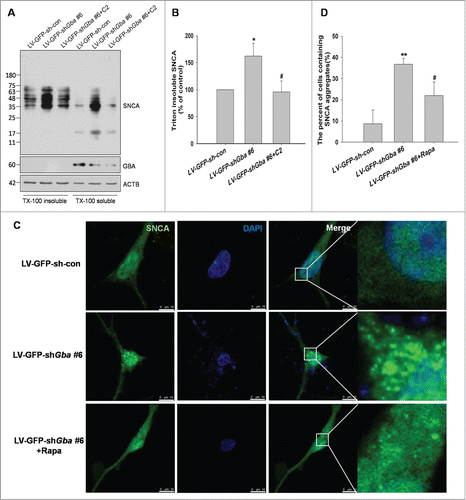
To confirm whether SNCA accumulation resulting from loss of GBA function was responsible for the inhibition of macroautophagy, the presence of high molecular weight SNCA species was examined by western blotting. Monomers of SNCA are recognized by the ubiquitin-proteasome system and chaperone-mediated autophagy and subsequently degraded. However, macroautophagy is the only mechanism for clearance of oligomers and SNCA aggregates.Citation29,30 We used solubility in 1% TritonX-100 to separate soluble from insoluble forms. The accumulation of high molecular weight SNCA species was significantly increased in the TritonX-100 insoluble fraction in LV-GFP-shGba #6-infected neurons, and this effect was abolished by treatment with C2 (). Furthermore, SNCA aggregation was observed in cortical neurons upon GBA knockdown, which was alleviated upon treatment with rapamycin ().These results provide evidence that GBA deficiency and consequent macroautophagic inhibition lead to the production and aggregation of SNCA oligomeric species.
Figure 10. SNCA oligomerization and aggregation by loss of GBA function. (A, B) The accumulation of high-molecular-weight SNCA species were significantly increased in the TritonX-100 insoluble fraction in LV-GFP-shGba #6-infected neurons, and this effect was abolished by treatment with C2 (5 μM for 8 h). (C) Neurons were infected with LV-GFP-shGba #6 or LV-GFP-sh-con for 3 d, and SNCA aggregation was assessed by thioflavin-S. Nuclei were stained with DAPI. Scale bar = 10 μm. (D) The number of cells containing SNCA aggregates was quantified in 6 randomly chosen microscopic fields. SNCA aggregation was observed in cortical neurons upon GBA knockdown, which was alleviated upon treatment with rapamycin (40 nM for 6 h). *P< 0.05, **P< 0.01 vs. LV-GFP-sh-con group, #P< 0.05, vs. LV-GFP-shGba #6 group; n = 3.
SNCA overexpression leads to the downregulation of GBA protein in SK-N-SH cells and cortical neurons and transgenic mice
An elevated level of SNCA protein is observed in most idiopathic PD patients; thus, the effect of SNCA overexpression was examined in SK-N-SH cells and cortical neurons. GBA protein level was downregulated in SNCA-overexpressing cells and neurons relative to control cells; moreover, PPP2A activity and LC3-II level were also reduced (Fig. S6), likely as a consequence of GBA deficiency. Transgenic mice overexpressing HsSNCA were examined in order to assess the level of autophagy and further confirm the relationship between GBA and SNCA. These results indicated that GBA protein level and LC3-II level were significantly decreased in transgenic mice compared to wild-type mice (Fig. S7A). In order to further assess whether PPP2A or autophagy inhibition by GBA knockdown contributes to neurodegeneration, cell viability was evaluated using the MTS and LDH release assays. GBA knockdown resulted in a decreased in cell viability and an increased in LDH release, compared to control neurons, which were reversed by application of C2 or rapamycin (Fig. S7B, C).These alterations in PPP2A activity or autophagy level or GBA and SNCA protein could contribute to neurodegeneration.
Discussion
The current study analyzed the contribution of the autophagy pathway to the SNCA accumulation induced by GBA deficiency in SK-N-SH cells, primary rat cortical neurons, and the rat striatum. The results revealed that the regulation of PPP2A activity by phosphorylation on Tyr307 mediates the inhibition of autophagic activity upon loss of GBA function.
Recent studies have shown an association between GBA variants and familial and sporadic PD cases.Citation5,31 Indeed, GBA mutations are currently the most common genetic risk factor associated with PD, and patients have 5 times greater likelihood of carrying mutations in GBA.Citation6 However, the mechanistic basis of this association is unknown. Deposits of SNCA have been identified in pathological aggregates, such as LBs, Lewy neurites, and oligodendroglial inclusions in PD and several other neurodegenerative disorders.Citation32 Here, it was shown that loss of GBA function caused a significant increase SNCA protein accumulation (), consistent with some previous reports;Citation16,17 other studies have found that suppression of GBA activity using the pharmacological inhibitor conduritol B epoxide has no effect on SNCA metabolism,Citation33 possibly because SNCA accumulation is GBA mutant-specific.Citation17 However, SNCA mRNA level was unaltered by GBA knockdown, suggesting that inadequate degradation of SNCA protein accounted for the elevated expression.
Autophagy is reportedly impaired in the brains of PD patients.Citation34 Additionally, expression of regulatory components of the autophagy-lysosomal pathway is altered in the temporal cortex of LBD patients.Citation35 In this study, a downregulation of macroautophagy markers such as BECN1 and LC3 was observed in GBA-deficient neuroblastoma cells and cortical neurons and in the striatum (). This is in accordance with the reduction in autophagy markers ATG5, ATG12, and LC3 in cultured neurons seen in a type II GD mouse model, in which application of the autophagy-lysosomal pathway inhibitor BafA1 caused a small but significant increase in LC3-II level.Citation36 A decreased LC3-II level compared to controls was actually observed upon GBA knockdown (), which was rescued by treatment with rapamycin. Constitutive expression of BECN1 induces macroautophagy, and in neurons overexpressing SNCA, as well as in brains of SNCA transgenic mice, elevated levels of BECN1 expression result in autophagy, lysosomal activation, decreased SNCA accumulation, and reduced synaptic and dendritic pathology.Citation18 BECN1 is bound to the anti-apoptotic protein BCL2 in the cytoplasm, and its dissociation from BCL2 is essential for the initiation of autophagosome formation.Citation37 Therefore, the reduced expression of BECN1 in GBA knockdown cells and tissues implies that autophagosome generation was impaired. Ceramide, a product of GBA activity, stimulates the expression of BECN1;Citation26 the observed downregulation of BECN1 expression could therefore be caused by a GBA knockdown-induced reduction in GBA expression and activity, and the consequent decrease in ceramide production (). Reduced GBA protein expression and activity are detected in the brains of PD patients carrying GBA mutations as well as in sporadic cases;Citation15 in the latter, ceramide is reduced in brain regions that accumulate SNCA and exhibit GBA deficiency.Citation38
The results of this study present the first evidence that the mechanistic link between GBA deficiency and inhibition of autophagy is provided by PPP2A. PPP2A plays a crucial role in the pathogenesis of neurodegenerative diseases.Citation39 For instance, PPP2A downregulation is thought to contribute to MAPT/tau hyperphosphorylation and aggregation, as well as amyloid-β production in AD.Citation28 Loss of GBA function led to an increase in PPP2A phosphorylation at Tyr307, accompanied by a suppression of PPP2A () and autophagic activities (), consistent with the recent finding that blockade of PPP2A, either pharmacologically by OA or by shRNA-mediated silencing of PPP2C, inhibits basal autophagy,Citation40 whereas C2-dependent PPP2A activation may induce the GBA knockdown-mediated autophagy pathway (). These results indicate that GBA knockdown inhibits autophagy via PPP2A inactivation, which is likely important for autophagosome formation (). This may be associated with a decrease in ceramide production (), which is a known inducer of autophagy.Citation26 Alternatively, it has been suggested that ceramide, a potent apoptotic agent, promotes the dephosphorylation of the antiapoptotic molecule BCL2 through activation of PPP2A,Citation41 in contrast with the results of the present study. These contradictory findings may be explained by the dose-dependence of ceramide action; for instance, 3 μM ceramide promotes neuronal growth, and apoptosis is induced only when the concentration exceeds 10 μM.Citation42 However, further studies are required to delineate the precise mechanism by which GBA deficiency leads to the hyperphosphorylation and inactivation of PPP2A.
PPP2A activity is regulated by 3 processes: phosphorylation, methylation, and the binding of endogenous inhibitors such as inhibitor-1 and -2 of PPP2A.Citation43,44 PPP2C phosphorylation at Tyr307 decreases PPP2A activity by 90% in vitro,Citation19 and is increased by the loss of GBA function. To determine the mechanism of PPP2A inactivation, the methylation status of PPP2A was examined. The methylation of the C subunit is required for the assembly of B55-type subunits,Citation45,46 and a decrease in PPP2A methylation and PPP2R2A/B55α expression is detected in the frontal and temporal cortices of AD patient brains.Citation47,48 The level of methylated PPP2A, representing the active form of the enzyme, was decreased in SK-N-SH cells and cortical neurons upon GBA knockdown (Fig. S8), indicating a suppression of PPP2A activity. A decrease in the expression of the B subunit was also observed in these cells, which likely led to reduced methylation of the C subunit. However, no changes were detected in the level of the A subunit (Fig. S8). As noted earlier, PPP2R5A/B56α and not the A subunit is upregulated by ceramide in REH cells;Citation41 thus, the decrease in PPP2C methylation and downregulation of PPP2R could further contribute to PPP2A inactivation induced by loss of GBA function.
In addition to the lysosome, autophagic vacuoles are a compartment required for SNCA clearance.Citation49 Moreover, gene replacement of BECN1, which stimulates autophagy, abrogates the deleterious effects of SNCA overexpression in animal models of PD and LBD.Citation18 Conversely, the oligomerization of SNCA is induced by suppression of autophagic activity.Citation50 GBA-deficient cortical neurons and tissue had increased levels of monomeric and oligomeric SNCA species, which was reversed by treatment with C2 or rapamycin, likely through upregulation of the macroautophagic pathway (). However, the contribution of the ubiquitin-proteasome protein degradation pathway to the clearance of SNCA cannot be overlooked.
Taken together, these data suggest that the loss of GBA function leads to SNCA accumulation through inhibition of the autophagy pathway via PPP2A inactivation; SNCA overexpression leads to a decrease in GBA protein, which may in turn promote SNCA accumulation and oligomerization in a positive feedback loop, ultimately resulting in neurodegeneration. This can explain the observation that in GD and sporadic PD models, decreased GBA activity causes abnormal SNCA accumulation, which consequently inhibits normal GBA function.Citation38,51, 52
In conclusion, this study demonstrated that loss of GBA function inhibits the autophagy pathway, through negative regulation of PPP2A activity and possibly some upstream components, resulting in SNCA accumulation. These results offer insight into the molecular basis of PD and other neurodegenerative diseases, and potentially provide novel targets for pharmacological treatment strategies.
Materials and Methods
Plasmid constructs and lentivirus (LV)
ShRNA (shGBA #9847–1, 5′-AGCTAAATATGTTCATGGCAT-3′; shGBA #9848–1, 5′-GACTTCATTGCCCGTGACCTA-3′) targeting a specific region of human GBA mRNA (NM_001005741.2), and a scrambled negative control (sh-con, 5′-TTC TCC GAA CGT GTC ACG T-3′), were cloned into the GV248 vector (Genechem, Shanghai, China).LV gene transfer vectors encoding GFP-shGBA #9848–1 (LV-GFP-shGBA #9848–1) and a scrambled shRNA used as a negative control (LV-GFP-sh-con) were synthesized by Genechem.
LV gene transfer vectors encoding GFP-shRNAs (LV-GFP-shGba #5, 5′-TAGCGAAGGCATTGAGTATAA-3′; LV-GFP-shGba #6, 5′-CAGGCCATATCTTGGGCATAT-3′; LV-GFP-shGba #7, 5′-GAGGCCTGGGCCAATTACTTT-3′), targeting specific regions of rat Gba mRNA (NM_001127639.1), a scrambled negative control (LV-GFP-sh-con, 5′-TTC TCC GAA CGT GTC ACG T-3′), a LV vector encoding human GBA (LV-GFP-HsGBA), and a LV vector encoding cherry-shRNA (LV-cherry-shAtg5, 5′-AGAAGATGTTAGTGAGATT-3′), targeting specific regions of rat Atg5 mRNA (NM_001014250) were constructed (Genechem). The plasmids and lentivirus used in this study are listed in .
Table 1 ShRNA-plasmids and shRNA-lentivirus used in this study
The human wild-type SNCA (HsSNCA) cDNA was obtained by reverse transcriptase PCR from human brain RNA. Primers 5′-CTGGAAGATATGCCTGTGGATC-3′ and 5′-AGCACTTGTACAGGATGGAAC-3′ were designed according to the SNCA sequence in GenBank (NM_000345.3). The cDNA was directionally cloned into the pCMV-myc plasmid (kindly provided by Dr. ZhiqingXu, Capital Medical University, China) and the orientation was verified by sequencing. An LV gene transfer vector encoding a GFP-HsSNCA fusion protein (LV-GFP-SNCA) was generated by Genechem.
Cell cultures and transfection
Human SK-N-SH cells
Human SK-N-SH neuroblastoma cells obtained from the American Type Culture Collection (HTB-11™) were grown in Dulbecco's Modified Eagle's Medium supplemented with 10% fetal bovine serum (Gibco, 10099–141) until 70% to 80% confluence, then transfected with plasmids using Lipofectamine 2000 (Invitrogen,11668019) according to the manufacturer's instructions. Cells were incubated at 37°C in a humidified atmosphere of 5% CO2 for the indicated times.
Primary rat cortical neurons
All experiments were approved by the Institutional Animal Care and Use Committee of Capital Medical University of Science and Technology (approval No. SCXK-2011–004) and performed in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Surgeries were performed under chloral hydrate anesthesia. Primary cortical neurons were prepared from Sprague-Dawley rat E14.5 to 15.5 embryos. Briefly, dissociated neurons were cultured in 6-well or 24-well plates on coverslips coated with 100 μg/mL poly-L-lysine (Sigma, P1524), in Neurobasal medium (Gibco, 21103–049) supplemented with L-glutamine (0.5 mM) and B27 supplement 50× (at a final concentration of 1×; Gibco, 17504–044). After 7 days, primary neurons were infected with LV.
Preparation of GBA knockdown rats by LV infection
The animal protocol was approved by the Animal Care and Use Committee of Capital Medical University and was consistent with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals (Publication No. 80–23). Male Sprague-Dawley rats (190 to 210 g) were housed under a 12:12 h light/dark cycle at 20°C to 23°C with free access to food and water. Rats were anesthetized with 6% chloral hydrate (6 ml/kg by intraperitoneal injection). Stereotaxic coordinates for injection into the striatum were: bregma anteroposterior 0 mm; lateral ±3 mm; and ventral −5 mm from the dura, with the tooth bar set at 0 mm. Suspensions of GFP-labeled LV vector (3×108 TU) in a 5-μl volume were injected into the left striatum of rats. At the end of the injection, the needle was left in place for 10 min before being slowly withdrawn. The skin was sutured and rats were allowed to recover. One wk after infection, rats were used for subsequent experiments.
Transgenic mice
The animal protocol was approved by the Animal Care and Use Committee of Capital Medical University and was consistent with the NIH Guide for the Care and Use of Laboratory Animals (NIH Publication No. 80–23). Male transgenic mice (18 to 22 g) overexpressing HsSNCA (TG) were purchased from the Jackson Laboratory (Bar Harbor, Maine, USA) numbered 017682, and maintained on a C57BL/6N background. Transgenic mice and wild-type (WT) littermates were housed under a 12:12 h light/dark cycle at 20°C to 23°C with free access to food and water.
Sample preparation and western blot analysis
After transfection or infection, cells or tissue were washed with ice-cold phosphate-buffered saline (PBS; Origene, ZLI-9062) and lysed in RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM sodium chloride, 1% NP-40 [Sigma, 74385], 0.1% SDS [Sigma, L5750]) containing phosphate and protease inhibitor cocktails (Roche, 04693132001). Homogenates were centrifuged at 12,000×g for 20 min and the supernatant fraction was collected for analysis. The protein concentration was determined using a bicinchoninic acid protein assay kit (Pierce Biotechnology, 23227) according to the manufacturer's instructions. A total of 20 μg of protein was resolved on 10% SDS polyacrylamide gels and transferred to PVDF membranes (Millpore, ISEQ00010). After blocking with 10% milk for 1 h, membranes were incubated with the following primary antibodies: rabbit anti-ACTB (1:5,000; Sigma, A5060), rabbit anti-ATG5 (1:1000; Cell Signaling Technology, 8540), rabbit anti-BECN1 (1:1,000; Proteintech Group, 11306–1-AP), rabbit anti-GBA (1:1,000; Epitomics, 5719–1), rabbit anti-LC3 (1:1,000; Novus Biologicals Ltd., NB100–2220), rabbit anti-phospho (p)-PPP2C (Tyr307) (1:1,000; Abcam, ab32104), mouse anti-PPP2A (1:1,000; BD Transduction Laboratories, 610556), mouse anti-methyl-PPP2C (1:500; Millipore, 04–1479), rabbit anti-PPP2R1 (1:1,000; Millipore, 07–250), mouse anti-PPP2R (1:1,000; Millipore, 05–592), rabbit anti-SQSTM1/p62 (1:1000; Cell Signaling Technology, 8025), mouse anti-3D5-SNCA (1:500; gift from Dr. ShunYu, Xuanwu Hospital, Capital Medical University, China), and mouse anti-SNCA (1:500; BD Transduction Laboratories, 610787). HRP-conjugated secondary antibodies (1:10,000) were purchased from KPL (04–18–06; 04–15–06). ACTB was used as a loading control. Immunoblots were visualized with super enhanced chemiluminescence detection reagent (Applygen, P1020) using a Gel-Doc 2000 imaging system (Bio-Rad, Hercules, USA).
SNCA aggregation assay
Primary rat cortical neurons were resuspended in 5 volumes of ice-cold TritonX-100 lysis buffer (50 mM Tris, pH 7.4, 175 mM NaCl, 5 mM EDTA, 1% TritonX-100 [Sigma, V900502], 1 mM PMSF), and incubated on ice for 30 min. Samples were centrifuged at 100,000 ×g for 30 min at 4°C and the resulting supernatant fraction was deemed the TritonX-100-soluble fraction. The pellet was resuspended in lysis buffer containing 2% SDS and following sonication on ice was designated as the TritonX-100-insoluble fraction.
RNA isolation and real-time PCR (RT-PCR)
Total RNA was isolated from SK-N-SH cells or tissue lysed in1 mL TRIzol reagent (Invitrogen, AM9738) in a 35-mm diameter dish, according to the recommended protocol. Cell or tissues lysates were transferred to tubes and 0.2mL of chloroform was added for total RNA extraction. Samples were centrifuged at 12,000×g for 15min at 4°C. RNA in the colorless, upper aqueous phase was transferred to a fresh tube where the RNA was precipitated by addition of isopropyl alcohol and centrifugation at 12,000×g for 10 min at 4°C. The supernatant was removed and the RNA pellet was washed once with 75% ethanol, dried for 10 min, and dissolved in RNase-free water.
The RNA was used to generate cDNA by reverse transcription using the Transcriptor High Fidelity cDNA Synthesis Kit (Roche, 05081955001) according to the manufacturer's instructions. The amount of cDNA was quantified by real-time PCR in a 7300 real-time PCR thermal cycler (Applied Biosystems, Carisbad, USA) using the following primer/probe sets (human SNCA: Assay ID, Hs01103383_m1; human GBA: Assay ID, Hs00986836_g1; rat Snca: Assay ID, Rn00569821_m1; rat Gba: Assay ID, Rn01455381_m1) and normalized to ACTB levels (human ACTB: Assay ID, Hs01060665_g1; rat Actb: Assay ID, Rn01775763_g1). Fluorescently labeled TaqMan primer/probe sets were manufactured by Applied Biosystems.
Immunofluorescence and confocal microscopy
Primary rat cortical neurons and SK-N-SH cells were seeded on poly-l-lysine-coated coverslips in 24-well plates and infected with LV-GFP-sh-con, LV-GFP-shGba #6, or LV-GFP-shGBA #9848–1. After 3 d, cells were fixed with 4% paraformaldehyde for 30 min, washed in PBS, and permeabilized with 0.3% Triton X-100 in PBS for 10 min at room temperature. After blocking with 10% normal goat serum for 1 h, cells were incubated with rabbit anti-TUBB3/tubulin III (1:1,000; Abcam, ab7751), mouse anti-ceramide (1:100; Sigma, C8104), rabbit anti-GlcCer/glucosylceramide (1:50; Glycobiotech, RAS_0011), mouse anti-LC3 (1:100; MBL, M152–3), and rabbit anti-LAMP1 (1:200; sigma, L1418) antibody overnight at 4°C, followed by incubation with Alexa Fluor 594-conjugated (1:500; Life Technologies, A-21207 or A-21203) and/or Alexa Fluor 647-conjugated secondary antibody (1:500; Life Technologies, A-21244) for one h at room temperature. SNCA aggregates were stained with Thioflavin S (Sigma, T1892) as previously described.Citation53 Briefly, cells were incubated with 0.5% Thioflavin S for 8 min, then washed in 80% ethanol. Cell nuclei were visualized by counterstaining with DAPI (Sigma, D9542). Coverslips were mounted on slides with 70% glycerol, and the cells were imaged using a confocal microscope (Leica, TCS SP8, Solms, Germany).
Brain tissue sections were prepared as previously described.Citation54 Rat brains were cut into 20-μm-thick sections on a cryostat and following immunohistochemistry, were mounted on slides with 70% glycerol and imaged using a confocal microscope (Leica).
GBA activity assay
GBA activity was measured using the QuantiChrom β-glucosidase Assay Kit (BioAssay Systems, DBGD-100), according to the manufacturer's instructions. In this assay, р-nitrophenyl-β-d-glucopyranoside is hydrolyzed specifically by GBA to yield a yellow product, with the maximal absorbance at 405 nm directly proportional to enzymatic activity. Absorbance was measured at the start of the reaction (0 min) and again after 20 min using a Bio-Rad model 3550 microplate reader (Richmond, USA). GBA activity was calculated by the equation
Where OD20 and OD0are OD405nmvalues at 20 min and 0 min, respectively, and ODcalibrator and ODwater are OD405nmvalues of the calibrator and water, respectively, at 20 min.
PPP2A activity assay
PPP2A activity was measured as previously reported,Citation55 using the PPP2A Colorimetric Assay kit (GenMed Scientifics Inc. USA, GMS50042.3/GMS50042.4). This assay is based on the release of free phosphate from the dephosphorylation of RKpTIRR by endogenous PPP2A, which is detected by a chromogenic reaction with molybdenum blue produced by a ferrous sulfate reduction. The free phosphate concentration was measured at 660 nm using a spectrophotometer (Bio-Rad, USA). The phosphate concentration (μM/L) was converted to PPP2A activity/mg protein as described in the following sections.
Sample preparation
Following transfection or infection, 5×106 cells grown in 25-cm2 culture flasks or 500 mg rat striatal tissue were used for the PPP2A activity assay. The medium was discarded after C2-ceramide (C2) treatment and 3 mL precooled reagent A (GENMED Cleaning Solution) was used to wash the cells or tissue, which were resuspended in 3 mL precooled reagent A, and centrifuged at 300×g for 5 min at 4°C. The supernatant fraction was discarded, and 500 μL reagent B (GENMED Lysis Solution) was added to the pellet. The lysate was incubated on ice for 30 min with strong vortexing for 30 s every 10 min, then centrifuged at 16,000×g for 5 min at 4°C. A 500-μL volume of the supernatant fraction containing soluble protein was transferred to a tube, and protein concentration was determined using a Bradford kit (GenMed Scientifics, GMS30030.1). PPP2A activity was measured as detailed below.
Determination of sample background, and total and nonspecific activities
Free phosphate present in the sample prior to the PPP2A-mediated reaction (i.e., background level) was determined by mixing 20 μL cell or tissue lysate containing 100 μg protein with 100 μL reagent C and incubating for 20 min at 30°C. Reagents D (60 μL) and F (200 μL) were added and the mixture was incubated for 10 min at 37°C in the dark. Reagent E containing the PPP2A substrate RKpTIRR was omitted to measure pre-existing free phosphate only. The absorbance was measured at 660 nm. For determination of total free phosphate, samples were treated as described above, except that 20 μL reagent E (GENMED Reaction Solution) was added after Reagent C, and the mixture was incubated for 20 min at 30°C. Nonspecific activity was determined as described for total free phosphate, with the addition of the PPP2A inhibitor okadaic acid (OA) (10 μL reagent H).
Calculation of PP2A activity
Total PPP2A activity was measured as total free phosphate released by PPP2A dephosphorylation. The nonspecific PPP2A activity was measured as free phosphate released from other processes (i.e., nonspecific activity insensitive to a PPP2A inhibitor). Both parameters were measured as the amount of free phosphate above sample background levels. The specific PPP2A activity of the sample was the OA-sensitive fraction of the total, and was calculated according to the following equations.
Lipid extraction and high performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS)
Lipids were extracted from cells using the Bligh&Dyer protocol typically used for biological matrices.Citation56 Briefly, cells grown on 100-mm plates were scraped and washed with cold PBS, then centrifuged for collection. Cells were mixed with chloroform followed by methanol (2:1, v/v) and sonicated before 0.5 mL ddH2O was added and the mixture was transferred to a 15-mL centrifuge tube. After centrifugation, the lower phase was transferred by aspiration to a clean glass tube, and the solvent was evaporated under N2. Samples were stored at −20°C until analysis. Except for the high-speed centrifugation, glass products were used in the remaining steps.
Cell extracts were analyzed on a 1290 Infinity chromatograph combined with a 6490 Triple Quad LC/MS system (Agilent Technologies, Santa Clara, CA, USA). The column (at 25°C) was an SB-C18 3.0×100 mm with 1.8-μm particles (Agilent Technologies), and solvents A and B were ultrapure water (with 0.1% HCOOH and 5 mM NH4Ac) and 100% acetonitrile, respectively. A flow rate of 0.2 mL/min and injection volume of 2.0 μL were used. The C2 standard (Sigma, A7191) was detected in the multiple reaction monitoring mode by MS/MS with electrospray ionization in the positive ion mode. The following conditions were applied: gas temperature=200°C, gas flow=14.0 L/min, nebulizer=25 psi, sheath gas temperature=400°C, sheath gas flow=11.0 L/min, and collision energy=25. Parent ions (m/z 342) were subjected to collision-induced dissociation and generated the daughter ion sphingosine, a characteristic ceramide fragment (m/z 264). The standard was used to confirm the retention time. Calibration curves were generated from serial dilutions of the standard (0.005, 0.05, 0.1, 0.5, 1, and 5 μg/mL). Samples were detected and extracted ion peaks (342.0 > 264.0) were integrated for quantitative analysis. The data were further normalized to the protein content of each sample.
Preparation of cellular sections and transmission electron microscopy (TEM)
Ultrastructural analysis was performed on primary rat cortical neurons. The primary neurons cultured in different conditions, were fixed in suspension with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (Wako, 039–18165) for 2 h at 4°C, and sufficient washed in 0.1 M PBS, then postfixed in 1% osmium tetroxide in 0.1 M cacodylate buffer for 1 h at room temperature. The samples were then dehydrated in graded ethanol and embedded in epoxy resin. Ultrathin sections were collected on Formvar coated copper grids (Beijing Xinxing Braim Technology Co., Ltd, T11022), stained with uranyl acetate and lead citrate, and observed on transmission electron microscope (JEOL, JEM-2100, Japan).
Cell viability assay
Cell viability was determined using a CellTiter 96® AQueous One Solution Cell Proliferation (MTS) assay kit (Promega, G3580) according to the manufacturer's instruction. Toxicity was evaluated by LDH-Cytotoxicity Detection Kit (Roche, 04744926001), following the manufacturer's protocol.
Statistical analysis
Western blotting results were analyzed as previously described.Citation55 Relative optical density values for bands corresponding to GBA, ATG5, BECN1, SQSTM1, LC3-II, SNCA, PPP2R1, PPP2R, and PPP2C were normalized to the value for ACTB. The normalized ratio for the control group (untreated SK-N-SH cells or cortical neurons) was taken as 100%. PPP2A phosphorylation and methylation levels were expressed as p-PPP2A/PPP2A or methyl-PPP2A/PPP2A ratio, with the ratio for the untreated group taken as 100%. Similarly, for the GBA activity, PPP2A activity and cell viability assays, values for the control group were taken as 100%, and data for other groups are expressed as a percentage of this value. For relative mRNA level, cycle threshold (Ct) values of the target transcript were normalized to ACTB Ct values and plotted as % of control. To evaluate ceramide and GlcCer level, the mean intensity for ceramide and GlcCer was calculated as the ratio of integrated optical density to area. Results were presented as mean ± SEM for at least 3 independent experiments. Statistical analysis was performed using ANOVA followed by a Bonferroni post-hoc correction for multiple comparisons. Differences were considered significant at P< 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
1086055_GBA_deficiency_promotes inhibition_by_inactivated_PPP2A.docx
Download MS Word (6.4 MB)Funding
This work was supported by grants from the National Basic Research Program of China (2011CB504102, 2012CB722407), National Natural Science Foundation of China (81371398, 31100767, 31271136, and 81200995), Natural Science Foundation of Beijing (7131001), Scientific Research Key Program of Beijing Municipal Commission of Education (KZ201210025020), and the Project of Construction of Innovative Teams and Teacher Career Development for Universities and Colleges Under Beijing Municipality (IDHT20140514).
Related Research Data
References
- Thomas B, Beal MF. Parkinson disease. Hum Mol Genet 2007; 16 Spec No. Two:R183-94; PMID:17911161; http://dx.doi.org/10.1093/hmg/ddm159
- Devine MJ, Gwinn K, Singleton A, Hardy J. Parkinson disease and α-synuclein expression. Mov Disord 2011; 26:2160-8; PMID:21887711; http://dx.doi.org/10.1002/mds.23948
- Lesage S, Brice A. Parkinson disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet 2009; 18:R48-59; PMID:19297401; http://dx.doi.org/10.1093/hmg/ddp012
- Mata IF, Samii A, Schneer SH, Roberts JW, Griffith A, Leis BC, Schellenberg GD, Sidransky E, Bird TD, Leverenz JB, et al. Glucocerebrosidase gene mutations: a risk factor for Lewy body disorders. Arch Neurol 2008; 65:379-82; PMID:18332251; http://dx.doi.org/10.1001/archneurol.2007.68
- Mitsui J, Mizuta I, Toyoda A, Ashida R, Takahashi Y, Goto J, Fukuda Y, Date H, Iwata A, Yamamoto M, et al. Mutations for Gaucher disease confer high susceptibility to Parkinson disease. Arch Neurol 2009; 66:571-6; PMID:19433656; http://dx.doi.org/10.1001/archneurol.2009.72
- Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson disease. N Engl J Med 2009; 361:1651-61; PMID:19846850; http://dx.doi.org/10.1056/NEJMoa0901281
- Lesage S, Anheim M, Condroyer C, Pollak P, Durif F, Dupuits C, Viallet F, Lohmann E, Corvol JC, Honore A, et al. Large-scale screening of the Gaucher's disease-related glucocerebrosidase gene in Europeans with Parkinson disease. Hum Mol Genet 2011; 20:202-10; PMID:20947659; http://dx.doi.org/10.1093/hmg/ddq454
- Brady RO, Kanfer J, Shapiro D. The Metabolism of Glucocerebrosides. I. Purification and Properties of a Glucocerebroside-Cleaving Enzyme from Spleen Tissue. J Biol Chem 1965; 240:39-43; PMID:14253443
- Hruska KS, LaMarca ME, Scott CR, Sidransky E. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat 2008; 29:567-83; PMID:18338393; http://dx.doi.org/10.1002/humu.20676
- Halperin A, Elstein D, Zimran A. Increased incidence of Parkinson disease among relatives of patients with Gaucher disease. Blood Cells Mol Dis 2006; 36:426-8; PMID:16651014; http://dx.doi.org/10.1016/j.bcmd.2006.02.004
- Clark LN, Ross BM, Wang Y, Mejia-Santana H, Harris J, Louis ED, Cote LJ, Andrews H, Fahn S, Waters C, et al. Mutations in the glucocerebrosidase gene are associated with early-onset Parkinson disease. Neurology 2007; 69:1270-7; PMID:17875915; http://dx.doi.org/10.1212/01.wnl.0000276989.17578.02
- Nichols WC, Pankratz N, Marek DK, Pauciulo MW, Elsaesser VE, Halter CA, Rudolph A, Wojcieszek J, Pfeiffer RF, Foroud T. Mutations in GBA are associated with familial Parkinson disease susceptibility and age at onset. Neurology 2009; 72:310-6; PMID:18987351; http://dx.doi.org/10.1212/01.wnl.0000327823.81237.d1
- Anheim M, Elbaz A, Lesage S, Durr A, Condroyer C, Viallet F, Pollak P, Bonaiti B, Bonaiti-Pellie C, Brice A. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 2012; 78:417-20; PMID:22282650; http://dx.doi.org/10.1212/WNL.0b013e318245f476
- Wong K, Sidransky E, Verma A, Mixon T, Sandberg GD, Wakefield LK, Morrison A, Lwin A, Colegial C, Allman JM, et al. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol Genet Metab 2004; 82:192-207; PMID:15234332; http://dx.doi.org/10.1016/j.ymgme.2004.04.011
- Gegg ME, Burke D, Heales SJ, Cooper JM, Hardy J, Wood NW, Schapira AH. Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann Neurol 2012; 72:455-63; PMID:23034917; http://dx.doi.org/10.1002/ana.23614
- Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011; 146:37-52; PMID:21700325; http://dx.doi.org/10.1016/j.cell.2011.06.001
- Cullen V, Sardi SP, Ng J, Xu YH, Sun Y, Tomlinson JJ, Kolodziej P, Kahn I, Saftig P, Woulfe J, et al. Acid β-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter α-synuclein processing. Ann Neurol 2011; 69:940-53; PMID:21472771; http://dx.doi.org/10.1002/ana.22400
- Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, Adame A, Wyss-Coray T, Masliah E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in α-synuclein models of Parkinson and Lewy body diseases. J Neurosci 2009; 29:13578-88; PMID:19864570; http://dx.doi.org/10.1523/JNEUROSCI.4390-09.2009
- Chen J, Martin BL, Brautigan DL. Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science 1992; 257:1261-4; PMID:1325671; http://dx.doi.org/10.1126/science.1325671
- Pitre A, Davis N, Paul M, Orr AW, Skalli O. Synemin promotes AKT-dependent glioblastoma cell proliferation by antagonizing PP2A. Mol Biol Cell 2012; 23:1243-53; PMID:22337773; http://dx.doi.org/10.1091/mbc.E11-08-0685
- Kar S, Palit S, Ball WB, Das PK. Carnosic acid modulates Akt/IKK/NF-kappaB signaling by PP2A and induces intrinsic and extrinsic pathway mediated apoptosis in human prostate carcinoma PC-3 cells. Apoptosis 2012; 17:735-47; PMID:22453599; http://dx.doi.org/10.1007/s10495-012-0715-4
- Mitra A, Menezes ME, Pannell LK, Mulekar MS, Honkanen RE, Shevde LA, Samant RS. DNAJB6 chaperones PP2A mediated dephosphorylation of GSK3beta to downregulate β-catenin transcription target, osteopontin. Oncogene 2012; 31:4472-83; PMID:22266849; http://dx.doi.org/10.1038/onc.2011.623
- Yorimitsu T, He C, Wang K, Klionsky DJ. Tap42-associated protein phosphatase type 2A negatively regulates induction of autophagy. Autophagy 2009; 5:616-24; PMID:19223769; http://dx.doi.org/10.4161/auto.5.5.8091
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/10.4161/auto.19496
- Shacka JJ, Klocke BJ, Roth KA. Autophagy, bafilomycin and cell death: the “a-B-cs" of plecomacrolide-induced neuroprotection. Autophagy 2006; 2:228-30; PMID:16874105; http://dx.doi.org/10.4161/auto.2703
- Scarlatti F, Bauvy C, Ventruti A, Sala G, Cluzeaud F, Vandewalle A, Ghidoni R, Codogno P. Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J Biol Chem 2004; 279:18384-91; PMID:14970205; http://dx.doi.org/10.1074/jbc.M313561200
- Komatsu M, Ichimura Y. Physiological significance of selective degradation of p62 by autophagy. FEBS Lett 2010; 584:1374-8; PMID:20153326; http://dx.doi.org/10.1016/j.febslet.2010.02.017
- Sontag E, Nunbhakdi-Craig V, Sontag JM, Diaz-Arrastia R, Ogris E, Dayal S, Lentz SR, Arning E, Bottiglieri T. Protein phosphatase 2A methyltransferase links homocysteine metabolism with tau and amyloid precursor protein regulation. J Neurosci 2007; 27:2751-9; PMID:17360897; http://dx.doi.org/10.1523/JNEUROSCI.3316-06.2007
- Cook C, Stetler C, Petrucelli L. Disruption of protein quality control in Parkinson disease. Cold Spring Harb Perspect Med 2012; 2:a009423; PMID:22553500; http://dx.doi.org/10.1101/cshperspect.a009423
- Ding WX, Yin XM. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy 2008; 4:141-50; PMID:17986870; http://dx.doi.org/10.4161/auto.5190
- Neumann J, Bras J, Deas E, O'Sullivan SS, Parkkinen L, Lachmann RH, Li A, Holton J, Guerreiro R, Paudel R, et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson disease. Brain 2009; 132:1783-94; PMID:19286695; http://dx.doi.org/10.1093/brain/awp044
- Kahle PJ. α-Synucleinopathy models and human neuropathology: similarities and differences. Acta Neuropathol 2008; 115:87-95; PMID:17932682; http://dx.doi.org/10.1007/s00401-007-0302-x
- Dermentzaki G, Dimitriou E, Xilouri M, Michelakakis H, Stefanis L. Loss of β-glucocerebrosidase activity does not affect α-synuclein levels or lysosomal function in neuronal cells. PLoS One 2013; 8:e60674; PMID:23580063; http://dx.doi.org/10.1371/journal.pone.0060674
- Friedman LG, Lachenmayer ML, Wang J, He L, Poulose SM, Komatsu M, Holstein GR, Yue Z. Disrupted autophagy leads to dopaminergic axon and dendrite degeneration and promotes presynaptic accumulation of α-synuclein and LRRK2 in the brain. J Neurosci 2012; 32:7585-93; PMID:22649237; http://dx.doi.org/10.1523/JNEUROSCI.5809-11.2012
- Klucken J, Poehler AM, Ebrahimi-Fakhari D, Schneider J, Nuber S, Rockenstein E, Schlotzer-Schrehardt U, Hyman BT, McLean PJ, Masliah E, et al. Alpha-synuclein aggregation involves a bafilomycin A 1-sensitive autophagy pathway. Autophagy 2012; 8:754-66; PMID:22647715; http://dx.doi.org/10.4161/auto.19371
- Osellame LD, Rahim AA, Hargreaves IP, Gegg ME, Richard-Londt A, Brandner S, Waddington SN, Schapira AH, Duchen MR. Mitochondria and quality control defects in a mouse model of Gaucher disease–links to Parkinson disease. Cell Metab 2013; 17:941-53; PMID:23707074; http://dx.doi.org/10.1016/j.cmet.2013.04.014
- Maiuri MC, Criollo A, Kroemer G. Crosstalk between apoptosis and autophagy within the Beclin 1 interactome. EMBO J 2010; 29:515-6; PMID:20125189; http://dx.doi.org/10.1038/emboj.2009.377
- Murphy KE, Gysbers AM, Abbott SK, Tayebi N, Kim WS, Sidransky E, Cooper A, Garner B, Halliday GM. Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson disease. Brain 2014; 137:834-48; PMID:24477431; http://dx.doi.org/10.1093/brain/awt367
- Veeranna Yang DS, Lee JH, Vinod KY, Stavrides P, Amin ND, Pant HC, Nixon RA. Declining phosphatases underlie aging-related hyperphosphorylation of neurofilaments. Neurobiol Aging 2011; 32:2016-29; PMID:20031277; http://dx.doi.org/10.1016/j.neurobiolaging.2009.12.001
- Magnaudeix A, Wilson CM, Page G, Bauvy C, Codogno P, Leveque P, Labrousse F, Corre-Delage M, Yardin C, Terro F. PP2A blockade inhibits autophagy and causes intraneuronal accumulation of ubiquitinated proteins. Neurobiol Aging 2013; 34:770-90; PMID:22892312; http://dx.doi.org/10.1016/j.neurobiolaging.2012.06.026
- Ruvolo PP, Clark W, Mumby M, Gao F, May WS. A functional role for the B56 α-subunit of protein phosphatase 2A in ceramide-mediated regulation of Bcl2 phosphorylation status and function. J Biol Chem 2002; 277:22847-52; PMID:11929874; http://dx.doi.org/10.1074/jbc.M201830200
- Mitoma J, Ito M, Furuya S, Hirabayashi Y. Bipotential roles of ceramide in the growth of hippocampal neurons: promotion of cell survival and dendritic outgrowth in dose- and developmental stage-dependent manners. J Neurosci Res 1998; 51:712-22; PMID:9545085; http://dx.doi.org/10.1002/(SICI)1097-4547(19980315)51:6%3c712::AID-JNR5%3e3.0.CO;2-E
- Tsujio I, Zaidi T, Xu J, Kotula L, Grundke-Iqbal I, Iqbal K. Inhibitors of protein phosphatase-2A from human brain structures, immunocytological localization and activities towards dephosphorylation of the Alzheimer type hyperphosphorylated tau. FEBS Lett 2005; 579:363-72; PMID:15642345; http://dx.doi.org/10.1016/j.febslet.2004.11.097
- Chen S, Li B, Grundke-Iqbal I, Iqbal K. I1PP2A affects tau phosphorylation via association with the catalytic subunit of protein phosphatase 2A. J Biol Chem 2008; 283:10513-21; PMID:18245083; http://dx.doi.org/10.1074/jbc.M709852200
- Yu XX, Du X, Moreno CS, Green RE, Ogris E, Feng Q, Chou L, McQuoid MJ, Pallas DC. Methylation of the protein phosphatase 2A catalytic subunit is essential for association of Balpha regulatory subunit but not SG2NA, striatin, or polyomavirus middle tumor antigen. Mol Biol Cell 2001; 12:185-99; PMID:11160832; http://dx.doi.org/10.1091/mbc.12.1.185
- Janssens V, Longin S, Goris J. PP2A holoenzyme assembly: in cauda venenum (the sting is in the tail). Trends Biochem Sci 2008; 33:113-21; PMID:18291659; http://dx.doi.org/10.1016/j.tibs.2007.12.004
- Sontag E, Hladik C, Montgomery L, Luangpirom A, Mudrak I, Ogris E, White CL, 3rd. Downregulation of protein phosphatase 2A carboxyl methylation and methyltransferase may contribute to Alzheimer disease pathogenesis. J Neuropathol Exp Neurol 2004; 63:1080-91; PMID:15535135
- Sontag E, Luangpirom A, Hladik C, Mudrak I, Ogris E, Speciale S, White CL, 3rd. Altered expression levels of the protein phosphatase 2A ABalphaC enzyme are associated with Alzheimer disease pathology. J Neuropathol Exp Neurol 2004; 63:287-301; PMID:15099019
- Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem 2003; 278:25009-13; PMID:12719433; http://dx.doi.org/10.1074/jbc.M300227200
- Yu WH, Dorado B, Figueroa HY, Wang L, Planel E, Cookson MR, Clark LN, Duff KE. Metabolic activity determines efficacy of macroautophagic clearance of pathological oligomeric α-synuclein. Am J Pathol 2009; 175:736-47; PMID:19628769; http://dx.doi.org/10.2353/ajpath.2009.080928
- Sardi SP, Clarke J, Viel C, Chan M, Tamsett TJ, Treleaven CM, Bu J, Sweet L, Passini MA, Dodge JC, et al. Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc Natl Acad Sci U S A 2013; 110:3537-42; PMID:23297226; http://dx.doi.org/10.1073/pnas.1220464110
- Yap TL, Velayati A, Sidransky E, Lee JC. Membrane-bound α-synuclein interacts with glucocerebrosidase and inhibits enzyme activity. Mol Genet Metab 2013; 108:56-64; PMID:23266198; http://dx.doi.org/10.1016/j.ymgme.2012.11.010
- Ostrerova-Golts N, Petrucelli L, Hardy J, Lee JM, Farer M, Wolozin B. The A53T α-synuclein mutation increases iron-dependent aggregation and toxicity. J Neurosci 2000; 20:6048-54; PMID:10934254
- Zhu Y, Duan C, Lu L, Gao H, Zhao C, Yu S, Ueda K, Chan P, Yang H. α-Synuclein overexpression impairs mitochondrial function by associating with adenylate translocator. Int J Biochem Cell Biol 2011; 43:732-41; PMID:21310263; http://dx.doi.org/10.1016/j.biocel.2011.01.014
- Yang W, Wang X, Duan C, Lu L, Yang H. Alpha-synuclein overexpression increases phospho-protein phosphatase 2A levels via formation of calmodulin/Src complex. Neurochem Int 2013; 63:180-94; PMID:23796501; http://dx.doi.org/10.1016/j.neuint.2013.06.010
- Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 1957; 226:497-509; PMID:13428781