
ABSTRACT
Autophagy is a cellular recycling pathway, which in many cases, protects host cells from infections by degrading pathogens. However, uropathogenic Escherichia coli (UPEC), the predominant cause of urinary tract infections (UTIs), persist within the urinary tract epithelium (urothelium) by forming reservoirs within autophagosomes. Iron is a critical nutrient for both host and pathogen, and regulation of iron availability is a key host defense against pathogens. Iron homeostasis depends on the shuttling of iron-bound ferritin to the lysosome for recycling, a process termed ferritinophagy (a form of selective autophagy). Here, we demonstrate for the first time that UPEC shuttles with ferritin-bound iron into the autophagosomal and lysosomal compartments within the urothelium. Iron overload in urothelial cells induces ferritinophagy in an NCOA4-dependent manner causing increased iron availability for UPEC, triggering bacterial overproliferation and host cell death. Addition of even moderate levels of iron is sufficient to increase and prolong bacterial burden. Furthermore, we show that lysosomal damage due to iron overload is the specific mechanism causing host cell death. Significantly, we demonstrate that host cell death and bacterial burden can be reversed by inhibition of autophagy or inhibition of iron-regulatory proteins, or chelation of iron. Together, our findings suggest that UPEC persist in host cells by taking advantage of ferritinophagy. Thus, modulation of iron levels in the bladder may provide a therapeutic avenue to controlling UPEC persistence, epithelial cell death, and recurrent UTIs.
Introduction
Urinary tract infections (UTIs) afflict more than 150 million people worldwide,Citation1,2 and over 25% of UTIs recur, resulting in both additional morbidity and a growing number of antibiotic-resistant UTI bacterial strains.Citation2,3 We have previously shown that the principal UTI pathogen, uropathogenic E. coli (UPEC), persists within the urothelium in autophagosomes. These organelles form during autophagy, a pathway in which damaged organelles, proteins, and invading pathogens are broken down and recycled via fusion of autophagosomes with lysosomes. Although autophagy commonly acts in defense against intracellular pathogens, we reported that inhibition of autophagy significantly reduced UPEC persistence in urothelial cells in mice.Citation4,5 However, the mechanisms underlying the ability of UPEC to interact with the autophagy pathway to survive intracellularly in the urinary tract remain unknown. Here, we explored whether UPEC is able to persist in autophagosomes because, in addition to processing cellular wastes, these organelles function in cellular control of iron processing.
Most forms of life require iron; for example, iron-sulfur clusters are essential components of proteins in the electron transport chain in mitochondria. Indeed, pathogens must acquire iron to survive inside host cells.Citation6-9 In particular UPEC have evolved a complex mechanism to chelate iron via siderophores, and several studies have revealed that UPEC have more iron chelators than most bacterial pathogens.Citation7,9-13 Consistent with this, UPEC growth is impeded if the bacteria are prevented from acquiring iron.Citation6,9,14 However, eukaryotic cells have evolved elaborate methods of iron sequestration, storage, and release.Citation15-22 Cells can sequester iron from extracellular sources via TF (transferrin)-bound iron or bioavailable iron. Iron can be stored intracellularly in a nonbioavailable form by binding to ferritin. Release of iron from ferritin occurs within autophagosomes in a process termed ferritinophagy. This selective form of autophagy specifically depends on NCOA4 to traffic ferritin to the autophagosome for degradation.Citation21,23-25
Here, we explored the connection between host cell iron regulation, ferritinophagy, and UPEC survival in bladder epithelial cells. We report that ferritinophagy occurs in bladder epithelial cells and that excess iron increases intracellular UPEC growth in a dose-dependent manner and leads to host cell death. Increased autophagy activity increases UPEC growth and inhibition of this process prevents UPEC overgrowth and host cell death. Thus, our findings demonstrate that autophagy is critical for intracellular UPEC survival and persistence. These findings open new opportunities for developing therapeutic options to treat recurrent UTIs.
Results
Iron treatment induces ferritinophagy
To determine whether iron treatment induces autophagy in bladder epithelial cells, we treated 5637 bladder epithelial cells (hereafter referred to as BECs) with 250 µM ferric ammonium citrate (FAC, referred to hereafter as iron treatment) and immunoblotted for MAP1LC3/LC3, a marker of autophagic activity.Citation26 We selected 250 µM as a representation of iron overload that may occur due to hemochromatosis or severe liver disease in humans.Citation21,27,28 We observed an increased LC3-II to LC3-I ratio after iron treatment, indicating accumulation of autophagosomes (). Further, the level of TFRC/CD71, the transferrin receptor and a transporter of iron from the extracellular milieu to the cytosol, decreased and the intracellular level of ferritin increased, suggesting that iron treatment increased iron uptake and storage and autophagic activity in BECs () We confirmed that treatment of BECs with another form of iron, di-ferric iron bound to TF derived from human blood, also caused an increase in the LC3-II:LC3-I ratio (Fig. S1A). Additionally, we observed that primary bladder cells responded to iron similarly to BECs (Fig. S1B). To determine whether the increased LC3-II:LC3-I ratio was due to an overall increase in autophagy or a block in autophagosome turnover, we treated the cells with bafilomycin A1 (Baf) to inhibit autophagic flux.Citation26 Since Baf can potentially disrupt the endocytic trafficking of TF, we also used chloroquine Citation29 and found that the LC3-II:LC3-I ratio was higher in cells treated with iron and either Baf or chloroquine than in cells treated with Baf or chloroquine alone (). Next, to determine whether chelation of iron would inhibit the autophagic response, we treated BECs with iron and the iron chelator deferoxamine (DFO)Citation30 and assessed the LC3-II:LC3-I ratio by western blot. We found that the LC3-II:LC3-I ratio, ferritin and TFRC levels in cells treated with both iron and DFO was similar to those noted in untreated cells (). Since DFO could induce mitophagy by itself,Citation31 we sought to determine if BECs were exhibiting specific activation of autophagy and not mitophagy, we added chloroquine to BECs treated with iron and DFO or with iron or DFO alone. Western blotting for MAP1LC3/LC3, PINK1, PARK2/PARKIN, and MFN2 (mitofusin 2) expression (markers of mitophagy) demonstrates there is a negligible increase in the LC3-II:LC3-I ratio upon addition of DFO alone and PINK1 or PARK2 remain unaffected (Fig. S1C). Together, these data demonstrate that DFO reverses the effect of iron overload in BECs and specifically decreases ferritinophagy.
Figure 1. Iron overload induces ferritinophagy. (A) Western blot analysis to detect the indicated proteins (GAPDH served as the loading control) in BECs treated with 250 µM iron (FAC) for the indicated time periods. (B) Western blot analysis of BECs treated with iron with or without Baf. (C) Western blot analysis of BECs treated with iron with or without chloroquine. (D) Western blot analysis of the indicated proteins in BECs treated with iron with or without DFO for 24 h. (E) Representative fluorescence images of iron-treated or untreated BECs expressing EGFP-LC3B (green). Scale bar: 20 µm. (F) Representative image of BECs stained with ferrum and LysoTracker Red to detect iron and lysosomes, respectively, after 24 h of treatment. Scale bar: 20 µm. (G) Representative image of BECs stained with ferrum and LysoTracker Red to detect ferritin and lysosomes, respectively, after 24 h of treatment. Scale bar: 20 µm. (H) TEM of an autophagosome in a BEC treated with 250 µM iron. Scale bar: 500 nm.
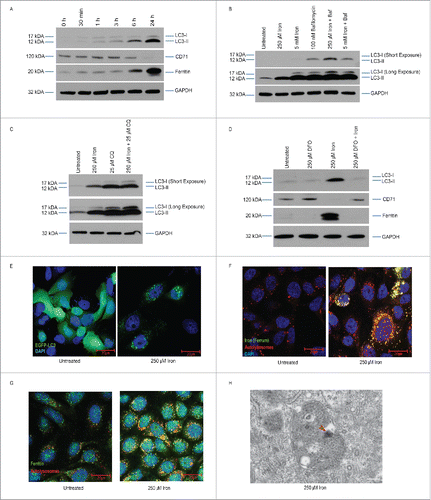
As an additional means of assessing the effect of iron treatment on autophagosome formation, we examined the formation of LC3-positive puncta in BECs transfected with an EGFP-LC3 plasmid. In untreated cells, MAP1LC3/LC3 appeared diffuse throughout the cytoplasm, but MAP1LC3/LC3-positive puncta were readily detectable in cells treated with iron (). If iron were being processed by autophagy, we would expect to observe colocalization of iron with lysosomes. Thus, we stained BECs with the lysosomal marker LysoTracker Red and iron indicators, ferrum and Calcein AM and found that both colocalized with LysoTracker Red indicating that iron is localized to the autolysosomal compartment (; Fig. S1D). In addition, we found that BECs stained with ferritin had increased colocalization of ferritin within autolysosomes and lysosomes (). Finally, we performed transmission electron microscopy (TEM) analysis of BECs after iron treatment and observed numerous double-membraned autophagosomes containing small electron-dense cores consistent with iron deposition (). Together, our findings show that iron treatment leads to ferritinophagy as indicated by increased autophagy and formation of iron-containing autophagosomes in BECs.
Iron treatment traffics UPEC into autophagosome and lysosome compartments
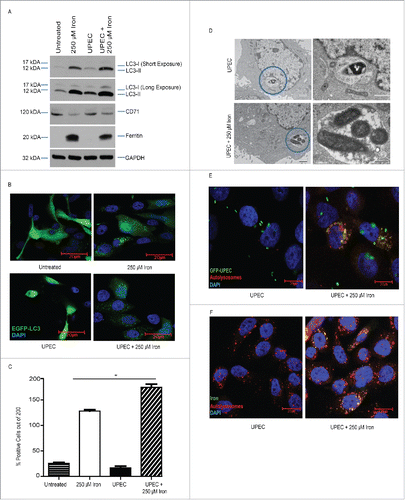
We have previously reported that UPEC can be taken up and persist within autophagosomes as quiescent intracellular reservoirs (QIRs) that can seed recurrent UTIs.Citation4,5,32 We reasoned that bacteria may form QIRs by trafficking together with ferritin-bound iron into the autophagosome. To test this idea, we challenged BECs with UPEC at a MOI of 100 with or without iron treatment and examined the effect on MAP1LC3/LC3 levels. We found that BECs treated with iron and UPEC had higher LC3-II levels and lower ferritin levels than BECs treated only with iron (). These effects were not observed in BECs challenged with the nonpathogenic E. coli strain MG1655 (Fig. S2A) or with UPEC alone in the absence of iron (). Immunofluorescence analysis confirmed that BECs treated with iron plus UPEC had more MAP1LC3/LC3 puncta than cells treated with either iron or UPEC alone (). Next, we examined the BECs by TEM analysis and found that in the absence of iron, UPEC were present as single organisms in autophagosomes, but in the presence of iron, multiple UPEC were seen in autophagosomes (). Furthermore, UPEC colocalized with lysosomes in the presence of iron (), and iron colocalized with lysosomes in the presence of UPEC (; Fig. S2B). Together, these findings suggest that UPEC shuttles with ferritin-bound iron into the autophagosome and/or lysosome compartments in BECs.
Figure 2. Iron traffics UPEC into autophagosomes. (A) Western blot analysis of the indicated proteins in BECs treated with or without iron during the 24 h UPEC infection. ((B)and C) Top, fluorescence micrographs of infected and iron-treated BECs transfected with EGFP-LC3B (green). Nuclei are stained with DAPI (blue). Bottom, quantification of LC3B puncta in BECs. Scale bar: 20 µm. (D) Representative TEM of BECs infected with UPEC in the absence (top) and presence (bottom) of iron. Images on the right depict details of autophagosomes. Circles indicate magnified area. Scale bar: 500 nm. (E) Fluorescence micrographs of infected and iron-treated BECs labeled with (E) GFP-UPEC (green) and LysoTracker Red and (F) ferrum (iron marker, green) and LysoTracker Red. Scale bar: 20 µm.
Iron treatment increases intracellular UPEC growth in BECs
Because UPEC efficiently chelate iron via their siderophores, we wondered whether iron treatment would enhance UPEC growth within BECs. We found that addition of iron led to a significant increase in intracellular bacterial load in BECs at both 6 h post infection (hpi) and 24 hpi (). We did not observe a significant difference in bacterial load at earlier time points (), suggesting that iron supplementation affected UPEC intracellular growth but not invasion. This response seemed to be specific to UPEC as intracellular growth of MG1655 was not altered by iron treatment ().
Figure 3. Iron increases intracellular UPEC growth. (A, B) Quantification of intracellular (A) UPEC and (B) MG1655 colony forming units (CFU) in BECs treated with or without 250 µM iron at the indicated hours postinfection (hpi). (C) Quantification of intracellular UPEC in BECs treated with 50 µM iron at 24 hpi. (D) Representative TEM image of an autophagosome in a UPEC-infected BEC treated with 50 µM iron. Scale bar: 500 nm. (E) Fluorescence micrograph of infected and iron-treated BECs stained with LysoTracker Red and labeled with GFP-UPEC (green). Scale bar: 20 µm. (F) Western blot of MAP1LC3/LC3, ferritin, and GAPDH (loading control) from BECs treated with the indicated iron concentrations for 24 h. (G) Quantification of intracellular UPEC in BECs treated with or without 50 µM iron for the indicated time periods.
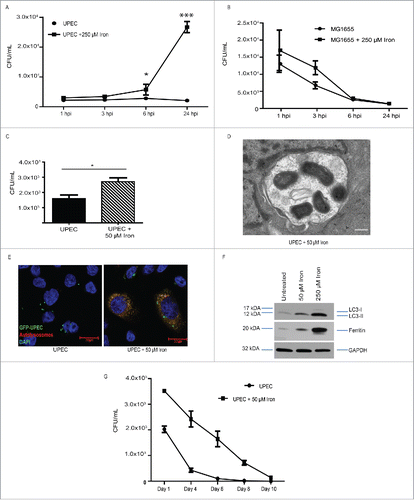
One concern we had was that LB broth contains iron (∼ 0.33 µM), which could contribute to UPEC growth in BECs. As expected, UPEC grown in minimal media supplemented with 250 µM FAC grew at a faster rate than bacteria in unsupplemented media (Fig. S3A). Next, we grew UPEC in minimal media or LB and then infected BECs. We found that the 2 UPEC preparations grew equivalently in BECs, and growth of both increased when the BECs were treated with iron (Fig. S3B), suggesting that UPEC efficiently uses iron in both nutrient-rich and minimal growth conditions. To confirm that treatment with the manufactured compound FAC was indicative of a host response to naturally available bio-iron, we infected BECs with UPEC in the presence of di-ferric iron bound to TF derived from human blood. UPEC grew to a similar extent in these cells as in FAC-treated cells (Fig. S3C). Finally, to confirm that this phenomenon was not unique to BECs, we infected primary bladder cells from the dome and apex regions of a normal human bladder in the presence or absence of iron.Citation33 In both cases, UPEC growth was enhanced in the presence of iron (Fig. S3D and E). Together, these data indicate that iron treatment promotes growth of UPEC in bladder cells.
To determine whether the high bacterial survival in BECs was due to the high level of iron (250 µM FAC), we treated BECs with a range of iron dosage from 10 µM to 250 µM. UPEC growth was significantly enhanced in as low as 25 µM iron (FAC) and 75 µM for Di-ferric TF doses (Fig. S3F and G). Even at 50 µM dose of iron, we observe increased bacterial burden in BECs (), find multiple UPEC clustered together in autophagosomes by TEM (), and note UPEC colocalizing with lysosomes (). Furthermore, BECs treated with 25 or 50 µM iron had higher LC3-II:LC3-I ratio (). At this range, iron treatment allowed bacteria to persist up to 8 d postinfection, at which point BECs that were not treated with iron had cleared their infections (; Fig. S3H). We conclude therefore that even physiologically relevant levels of iron are sufficient to support and prolong bacterial growth and survival in BECs.
Iron treatment increases host epithelial cell death upon UPEC infection due to lysosomal degradation
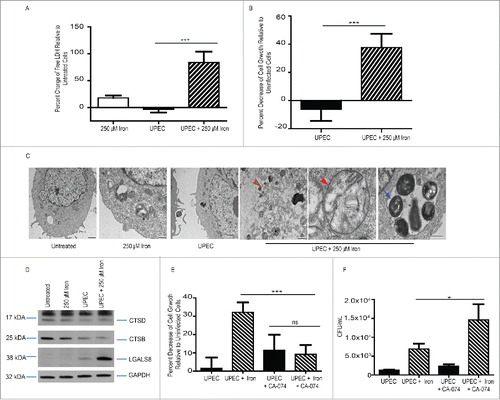
Iron overload is known to induce cell death in epithelial cells.Citation21,34-37 To assess the effect of iron and UPEC infection on BEC viability, we measured activity of LDH (lactate dehydrogenase) in media and performed cell growth assays at 24 hpi. We found that neither iron supplementation alone nor UPEC infection alone induced cell death, but the combination of iron and UPEC led to a significant increase in host cell death: approximately 40% of BECs infected with UPEC in the presence of 250 µM iron died by 24 hpi (; Fig. S4A). TEM analysis revealed permeabilized cells and deformed mitochondria in BECs infected with UPEC in the presence of iron (). In contrast, cells infected with MG1655 in the presence of iron did not die (Fig. S4B). However, cell death was dependent on iron dosage as no decrease in cell growth in BECs infected with UPEC was observed in the presence of 50 µM iron (Fig. S4C).
Figure 4. UPEC and iron-induced lysosomal damage leads to host cell death. (A) Relative percent change of LDH levels in BECs treated with 250 µM iron, infected with UPEC, or both. (B) Relative percent change in cellular growth of BECs infected with UPEC in the presence or absence of 250 µM iron. (C) Representative TEM images of BECs treated as indicated. The green arrow indicates loss of plasma membrane integrity (green arrow), the orange arrow indicates mitochondrial stress, and the blue arrow indicates multiple bacteria within an autophagosome. Scale bar: 500 nm. (D) Western blot analysis of CTSB and CTSD, LGALS8, and GAPDH in BECs treated as indicated. (E) Relative percent change in cell growth of BECs treated as indicated. (F) Quantification of bacterial burden in BECs treated as indicated.
We next sought to determine the mechanism underlying host cell death in the presence of UPEC and iron. We first considered apoptosis and inflammatory-mediated apoptosis, pyroptosis.Citation38 To assess apoptosis, we used western blots to measure levels of poly-ADP ribose polymerase (PARP1) and cleaved PARP1, which increases during apoptosis. However, we observed no evidence of cleaved PARP1 (Fig. S4D), indicating that iron supplementation did not lead to apoptosis. To examine pyroptosis, we measured levels of IL1B/interleukin 1β as we and others have shown that UPEC infection induces activation of IL1B in an NLRP3 inflammasome- and caspase 1-dependent manner in macrophagesCitation39,40 and epithelial cells.Citation41 Although UPEC infection led to an increase in the level of IL1B, the levels of IL1B, NLRP3, and Caspase 1 did not differ between cells infected with UPEC and those infected with UPEC in the presence of iron (Fig. S4D and data not shown). Together, these data indicate that increased cell death as a result of UPEC infection in the presence of iron was not due to canonical apoptosis or pyroptosis.
We wondered whether the cell death might be due to increased iron trafficking to lysosomes, which could lead to lysosomal permeability,Citation18 and examined levels of 3 markers of lysosomal damage, CTSB and CTSD and LGALS8/galectin-8.Citation42 Western blot analysis revealed that BECs treated with UPEC and iron had lower levels of CTSB and CTSD and higher levels of LGALS8 than untreated cells, cells treated with only UPEC, or cells treated with only iron, suggesting that there is increased lysosomal damage ().
To determine whether inhibition of lysosomal degradation could inhibit host cell death, we treated BECs with a CTSB inhibitor, CA-074. CA-074 treatment improved viability of BECs infected with UPEC in the presence of iron (). However, the increased survival of the BECs resulted in increased bacterial burden (). Together, these findings indicate that the cell death caused by UPEC plus iron was due to lysosomal damage.
Inhibition of the iron regulatory process reverses UPEC overgrowth and promotes host cell survival
Our results thus far demonstrated that iron treatment promoted intracellular UPEC survival and impaired host epithelial cell viability. Next, we sought to determine whether iron chelation could inhibit UPEC growth and promote host epithelial cell survival. Crystal violet cell growth assay revealed that iron chelation with DFO prevented BEC death wrought by iron treatment and UPEC infection (). In addition, DFO treatment reversed the increased bacterial growth () and the increased LC3-II:LC3-I ratio and ferritin levels () observed in cells treated with iron. Together, these data indicate that iron chelation improved host cell survival, impaired UPEC growth, and reduced autophagy.
Figure 5. Inhibition of the iron regulatory pathway reverses UPEC growth and iron-induced damage. (A) Relative percent change in cell growth of BECs treated as indicated. (B) Quantification of bacterial load in BECs treated as indicated. (C) Western blot of ferritin, LC3, and GAPDH (loading control) in BECs treated as indicated. (D) Western blot of ACO1, IREB2, LC3, ferritin, and GAPDH in BECs in which ACO1, IREB2, or both were knocked down. (E) Quantification of bacterial load in iron-treated, infected BECs in which ACO1, IREB2, or both were knocked down. (F) Relative percent change in cell growth of BECs in which ACO1, IREB2, or both were knocked down. (G) Western blot of ferritin, LC3, and GAPDH in BECs treated as indicated. (H) Relative percent change in growth of BECs challenged with UPEC in the presence of iron, OMA, or both.
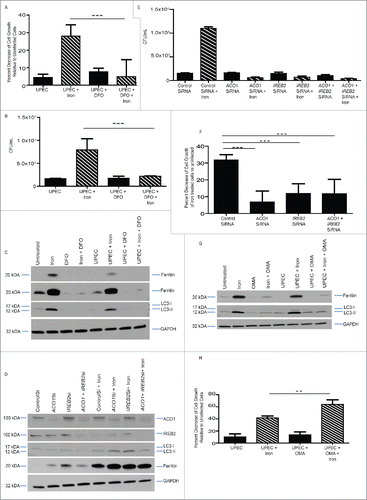
The iron storage and uptake is regulated by iron regulatory proteins, ACO1/IRP1 and IREB2/IRP2. Particularly, ACO1 is activated when it binds to iron-sulfur clusters. When iron is depleted and levels of iron-sulfur clusters are reduced, ACO1 binds to small hairpins on ferritin (FTH1 and FTL) mRNA to block its transcription, thus permitting levels of free iron to increase.Citation20,43 We used siRNAs to knock down expression of ACO1 and IREB2 proteins and found that their loss led to an increase in ferritin levels in uninfected cells (), suggesting that increased ferritin production promotes increased iron storage and sequestration away from UPEC. Indeed, knockdown of ACO1, IREB2, or both led to a significant reduction of bacterial burden () and host cell death (). In contrast, blocking the iron regulatory process by treating cells with oxalomalic acid (OMA), a potent inhibitor of ACO1 aconitase activity,Citation21 led to decreased ferritin levels by itself and upon iron treatment and UPEC infection (). Consistent with this reduction in ferritin levels, OMA treatment led to increased death of cells infected with UPEC in the presence of iron (), indicating that ferritin levels were critical to control intracellular bacterial burden. Together, our findings demonstrate that inhibiting the iron regulatory process promoted host cell survival and prevented UPEC overgrowth.
Inhibition of ferritinophagy inhibits UPEC overload and promotes host cell survival
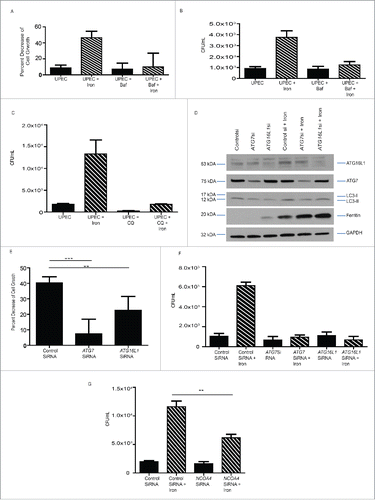
To determine whether inhibition of autophagy could protect BECs from cell death induced by UPEC infection in the presence of iron, we treated cells with chloroquine or Baf, which inhibits autophagosome-lysosome fusion. Treatment with either chloroquine or Baf reduced host cell death (; Fig. S5A) and UPEC growth (). Similarly, siRNA-mediated knockdown of the autophagy proteins ATG7 and ATG16L1 () reduced both host cell death () and bacterial burden () in BECs. Together, these findings suggest that canonical autophagy is critical for bacterial growth and host cell death observed under iron overload condition. Finally, we sought to demonstrate that UPEC growth in BECs is governed specifically via ferritinophagy. To do this, we knocked down NCOA4, a selective autophagy receptor for ferritin that traffics ferritin to autophagosomes for degradation.Citation23,24,44 We found that first, NCOA4 expression increases in cells exposed to iron () and second, loss of NCOA4 via siRNA knockdown in BECs results in reduction of bacterial burden induced by iron overload (; Fig S5C and D). Thus, our findings demonstrate that UPEC depend on NCOA4 expression and ferritinophagy to survive and persist in epithelial cells.
Figure 6. Ferritinophagy governs UPEC growth and cell survival. (A) Relative percent change in cell growth of BECs treated as indicated. ((B)and C) Quantification of bacterial load in BECs treated as indicated. (D) Western blot confirmation of ATG7 and ATG16L1 knockdown in BECs. (E) Relative percent change in cell growth of BECs treated as indicated. (F) Quantification of bacterial load in BECs in which ATG7 or ATG16L1 were knocked down. (G) Quantification of bacterial load in BECs in which NCOA4 was knocked down.
Discussion
In this report, we present several lines of evidence to support that UPEC persistence within bladder epithelial cells is facilitated by ferritinophagy. First, we show that iron treatment of urothelial cells induces ferritinophagy, a process in which ferritin-bound iron is taken up by autophagosomes and broken down to allow free iron to be released into the cytosol, making it available to the bacteria. Second, treatment with both moderate and high levels of iron increased autophagic flux and UPEC growth in urothelial cells. Third, ferritin, iron, and UPEC localized to autophagosomes and autolysosomes. Fourth, UPEC infection in the presence of iron overload induced host cell death due to lysosomal damage. Finally, selective inhibition of iron processing or ferritinophagy or iron chelation decreased bacterial growth in urothelial cells and reversed host cell death. Together, our findings suggest that ferritinophagy is co-opted by UPEC to survive and persist in bladder epithelial cells and modulation of this process can successfully impede UPEC survival. These data help explain the conundrum of why deficiency in an autophagy gene, ATG16L1, improves bacterial clearance during a urinary tract infection in mice.Citation4,5,39 We posit that in ATG16L1 deficiency, UPEC are unable to sequester iron and thus have a reduced ability to proliferate and persist in the bladder.
Our data highlight an intersection between iron regulation in epithelial cells and autophagy that may occur in many cell types. Although this is the first report of ferritinophagy in bladder cells, this pathway has been observed in ovarian, liver, and brain cell types.Citation21-24,44-48 In addition to observing an effect of iron on levels of autophagy, we also noted that loss of autophagy gene, ATG16L1 as well as selective autophagy receptor for ferritin that traffics ferritin to autophagosomes for degradation, NCOA4, resulted in increased levels of ferritin, suggesting that reduction of autophagy can enhance the ability of bladder cells to store iron. Because ferritinophagy can be utilized by pathogens, it must be carefully regulated. Our studies show that UPEC acquisition of intracellular iron is directly correlated to ferritinophagy and ferritin stores. When ferritin levels were reduced by genetically or pharmacologically inhibiting iron regulatory protein activity, UPEC growth increased while augmentation of ferritin levels blocked bacterial growth. Similarly loss of NCOA4 significantly reversed UPEC growth due to iron. Thus, our findings demonstrate that UPEC is unable to sequester iron from ferritin and must obtain free iron either via the cytosolic labile iron pool or free iron from degraded ferritin. This suggests that UPEC may directly or indirectly drive ferritinophagy by depleting the labile iron pool and forcing BECs to breakdown ferritin in order to provide iron for basic cellular functions.
UPEC is not the only bacteria capable of modulating host iron-processing pathways. For example, infections caused by the Dengue virus and Mycobacterium tuberculosis result in increased levels of ferritinCitation49,50 and increased host cell uptake of iron. Similarly, Chlamydia infection results in decreased ACO1 activity and thereby increased ferritin levels.Citation51 In contrast, visceral Leishmania infection results in increased levels of IREB2,Citation52,53 suggesting that pathogens can directly influence ferritin production and that IRPs have a strong influence over the fate of invading pathogens. Our studies reported here confirm that inhibiting IRP activity impedes UPEC overgrowth.
Iron dysregulation has clinically been noted in increased risk of infection both in patients with hemochromatosis or anemic patients receiving iron supplementation.Citation27,28,54,55 In both of these scenarios, excess free iron is present leading to opportunistic pathogenic burden. A number of case studies have even documented E. coli-associated bacteremia in hemochromatosis patients.Citation27,55 Iron overload is detrimental to the host not only due to supporting bacterial overgrowth, but also inducing epithelial cell stress. Indeed, we observed that the presence of high iron and UPEC infection and increased autophagy all resulted in host cell death. Mobley and colleagues have found that urine from patients with acute UTIs contain iron concentrations of 724 ± 185 nM which is significantly higher than those noted in healthy controls (161 ± 69 nM) and UTI urine samples contain significantly greater number of sloughed epithelial cells compared to urines from healthy subjects.Citation9,11,12 Furthermore, iron overload is generally considered to induce increased inflammatory activity.Citation15,17,56 We posit that when there is excess iron in the bladder or in the urine, this could augment inflammation and increase urothelial cell death in the course of infection.
Our findings that high iron leads to increased lysosomal degradation upon infection suggests that this may be relevant to understanding the impact of infection and iron in different cell types and diseases. Growing evidence suggests that there is a direct correlation between sensitivity to iron and lysosomal dysfunction in various diseases such as Alzheimer disease.Citation57 Disruption of a lysosomal membrane protein leads to iron accumulation in Kupffer cells in the liver resulting in spontaneous liver fibrosis;Citation58 ovarian cancer cells are sensitive to iron overload and die as a result.Citation21,22 Other studies have also linked high intralysosomal iron accumulation with oxidative stress and resulting cytotoxicityCitation34 consistent with a recent study showing that lysosomal integrity is necessary for UPEC to take advantage of excess bioavailable iron.Citation59
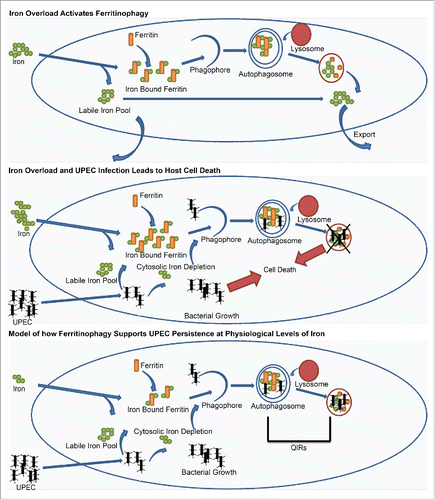
Normal serum iron levels in healthy adults are typically under 31 µMCitation60 and patients with severe hemochromatosis or liver damage can exhibit levels far greater than 31. Patients with iron dysfunction are more likely to experience severe bacterial burden which may result in damage to the bladder urothelium. This is particularly interesting because infants who tend to have higher basal iron serum levels or increased iron supplementation respectively are more at risk for UTIs.Citation54,61-63 Similarly, postmenopausal women who are highly susceptible to multiple recurrent and chronic UTIs have been shown to carry higher than normal levels of iron in their urine.Citation61,64,65 Given our findings that even at physiological levels of iron (25 µM) we observe increased and prolonged bacterial burden, our data shows that even slightly augmented levels can be beneficial for UPEC persistence. Together, our data support the model (illustrated in ) wherein high levels of iron increases ferritinophagy and leads to iron colocalizing in autophagosomes and autolysosomes (). UPEC infection in the presence of high iron results in increased bacterial burden and host cell death as a result of lysosomal damage (). In contrast, at physiological levels of iron host cell death does not occur, but UPEC are able to persist for prolonged periods. We thus propose that the quiescent intracellular reservoirs (QIRs) of UPEC found within autophagosomes in vivo, which can seed recurrent infections, are a result of UPEC following iron into autophagosomes where they are able to chelate iron via their siderophores (). Thus, modulation of iron levels and ferritinophagy could be of clinical benefit in reducing UPEC persistence and preventing UTI recurrence.
Figure 7. Model of ferritinophagy in UPEC pathogenesis. (A) Iron overload activates ferritinophagy (B). Iron overload and UPEC infection leads to host cell death (C). Model of how ferritinophagy supports UPEC persistence at physiological levels.
Materials and Methods
Cell lines
The human bladder carcinoma cell line 5637 (ATCC, HTB-9; referred to hereafter as bladder epithelial cells [BECs]) was maintained in RPMI medium (GIBCO, 11875-085) with 10% fetal bovine serum. The primary cell lines APEX and DOME (Lifeline Cell Technology, FC-0040 and FC-0079, respectively) were maintained in Urolife Media (Lifeline Cell Technologies, LL-0063 and LM-0042, res-pectively).
Bacterial strains and infections
A clinical UPEC isolate, UTI89,Citation39 or a nonpathogenic strain, MG1655Citation39 were grown statically for 17 h in Luria-Bertani (LB) broth at 37°C prior to infection of cells. Confluent BECs were challenged with bacteria (UPEC or MG1655) at a multiplicity of infection of 100. After bacteria were added, plates were centrifuged at 120 g for 5 min, and then incubated for 1 h at 37°C. Bacteria were then removed, BECs were washed twice with phosphate-buffered saline (PBS; Sigma-Aldrich, D8537), and medium containing 0.1 mg/mL gentamicinCitation66 was added to remove extracellular bacteria. BECs were incubated in gentamicin-containing medium for an additional 23 h (referred to as 24 h postinfection [hpi]) or the indicated number of hours.
Colony formation, lactose dehydrogenase (LDH) activity, and cell growth assays
Each of these assays was performed after the infection protocol described above. For the colony formation assay, BECs were washed at 1, 3, 6, or 24 hpi and treated with 0.1% Triton-X100 (Ricca Chemical Company, 8698.5–16) to release bacteria. Serial dilutions of bacteria were plated on LB agar, and colony forming units were counted. For the LDH assay, plates were centrifuged at 24 hpi, and media was collected and assayed with the LDH reaction kit (Promega, G1780) according to the manufacturer's suggested protocol. For the cell growth assay, at 24 hpi, BECs were stained with crystal violet solution for 10 min, washed, and allowed to dry overnight. Sorenson Buffer was added to dissolve stained cells, and levels were read on a plate reader at 590 nm.
Treatments with FAC, Di-ferric TF (transferrin) and inhibitors
BECs were treated with the indicated concentrations of ferric ammonium citrate (FAC; Sigma, F5879) dissolved in PBS, 250 µM (or 1:1 with the selected FAC concentration) deferoxamine (DFO; Sigma, D9533-1G) in PBS, 10 µM to 250 µM di-ferric iron (Lee Biosolutions, 535-11-100), 100 nM bafilomycin A1 (Baf) in DMSO, 25 µM chloroquine (Sigma, C6628), 5 mM oxalomalic acid (Cayman Chemicals, CAS89304-26-7), or 10 µM CTSB/cathepsin B inhibitor CA-0123 (Sigma, C5857) in DMSO. In experiments in which these compounds were added during infection (according to the protocol described above), the compounds were added during both the one-h bacterial infection and the 23-h gentamicin treatment period.
Transfection with siRNA and EGFP-LC3B
The TransIT-X2 (Mirus, MIR 6000) reagent was used to transfect cells with an EGFP-LC3 plasmid (Addgene, 11546 deposited by Karla Kirkegaard) or with siRNAs (Life Technologies) targeting ACO1/IRP1 (IDs224135), IREB2/IRP2) (IDs7498), ATG16L1 (IDs30071), ATG7 (IDs20651), NCOA4 (IDs107703) or nontargeting control siRNA (4390844).
Protein harvest, SDS-PAGE, and western blot analyses
At 24 hpi, cells were lysed with RIPA and cell lysates were electrophoresed on 4–20% Pierce Precise Protein Gels (Bio-Rad, 456-1094) and transferred to PVDF membranes. Primary antibodies were used at the following dilutions: LC3B rabbit polyclonal (1:1000; Novus, NB6000-1384), ATG7 rabbit polyclonal (1:1000; Sigma, A2856), ATG16L1 rabbit polyclonal (1:1000; Sigma, A7356), GAPDH rabbit polyclonal (1:2000; Cell Signaling Technology, 14C10), FTH1 (ferritin, heavy polypeptide 1) rabbit polyclonal (1:1000; Cell Signaling Technology, D1D4), TFRC/CD71 mouse monoclonal (1:100; Santa Cruz Biotechnology, sc-393719), ACO1 (1:500; Abcam, ab126595), cleaved PARP1 rabbit polyclonal (1:1000; Cell Signaling Technology, 5625), IREB2 (1:500; Abcam, ab80339), NCOA4 (1:200; Santa Cruz Biotechnology, sc-373739), PINK1 (1:250; Abcam, ab75487), PARK2/PARKIN (1:250; Abcam, ab77924), MFN2/mitofusin-2 (1:500; Cell Signaling Technology, 11925) CTSB/cathepsin B (1:250; Santa Cruz Biotechnology, G60), CTSD/cathepsin D (1:500; Santa Cruz Biotechnology, sc-6486), LGALS8/Galectin-8 (1:500; Santa Cruz Biotechnology, sc-28254), and IL1B/IL-1beta (1:1000; Santa Cruz Biotechnology, sc-1250).
LysoTracker Red, Calcein AM, ferritin, and ferrum staining
Media was prewarmed at 37°C, room temperature LysoTracker Red (Life Technologies, L-7528) was added to a concentration of 75 nM, and the media was added to cells for the last one h of the 24 h infection protocol. For Calcein AM staining, 1 ml of 0.15 μM Calcein AM (Invitrogen, C3100MP) in PBS containing 1 mg/ml BSA (Sigma, A9647) and 20 mM HEPES was added to the cells for 15 min at 37°C after bacterial challenge and gentamicin treatment. Cells were treated with a FTH1 (ferritin, heavy polypeptide 1) antibody (1:250; Santa Cruz Biotechnology, sc-25617) and stained with Alexa Fluor 488 (1:500; ThermoFisher, z-25302). Ferrum (Ursa Bioscience, 520-R) was dissolved in chloroform at room temperature and added to live cells at a 1:400 dose in complete media. Cells were treated for 1 h at 37°C. Cells were imaged with a Leica TSC SPE inverted confocal microscope (Wetzlar, Germany) and images were captured at 63x magnification with Leica Application Suite X software. Individual images for each channel are included in Figure S6.
Transmission electron microscopy (TEM)
BECs were fixed at 24 hpi in 2% paraformaldehyde/2.5% glutaraldehyde (Polysciences Inc.) in 100 mM cacodylate buffer, pH 7.2, for 1 h at room temperature as described.Citation39 Samples were washed in cacodylate buffer, postfixed in 1% osmium tetroxide (Polysciences Inc., 0972B-5) for 1 h, rinsed extensively in dH20, stained with 1% aqueous uranyl acetate (Ted Pella, 19481) for 1 h, rinsed several times in dH20, dehydrated in a graded series of ethanol, and embedded in Eponate 12 resin (Ted Pella, 18028). Sections (95 nm) were cut with a Leica Ultracut UCT ultramicrotome (Leica Microsystems Inc., Bannockburn, IL), stained with uranyl acetate and lead citrate, and viewed on a JEOL 1200 EX transmission electron microscope (JEOL USA Inc., Peabody, MA) equipped with an AMT 8-megapixel digital camera (Advanced Microscopy Techniques, Woburn MA).
Statistical analyses
All experiments were carried out with n = 3 replicates, except cell growth assays which were n=6 replicates. Graphpad Prism 5.0 software was used for statistical analysis. Error bars indicate standard deviation, and P-values were calculated with a standard Student t test. NS, not significant (P > 0.05); *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Abbreviations
| ACO1/IRP | = | aconitase 1 |
| Baf | = | bafilomycin A1 |
| CTSB | = | cathepsin B |
| CTSD | = | cathepsin D |
| DAPI | = | 4′,6-diamidino-2-phenylindole |
| EGFP | = | enhanced green fluorescent protein |
| FAC | = | ferric ammonium citrate |
| LGALS8/galectin-8 | = | lectin, galactoside-binding, soluble, 8 |
| IRE | = | iron-responsive element |
| IREB2 | = | iron responsive element binding protein 2 |
| MFN2 | = | mitofusin 2 |
| PBS | = | phosphate-buffered saline |
| QIR | = | quiescent intracellular reservoir |
| TEM | = | transmission electron microscopy |
| TFRC/CD71 | = | transferrin receptor |
| UTIs | = | urinary tract infections |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
1160176_Supplemental_Material.zip
Download Zip (13.2 MB)Acknowledgments
We thank Dr. Wandy Beatty, director of the Imaging Facility, Department of Molecular Microbiology, for TEM assistance. We thank Dr. Caihong Wang for technical advice and Drs. Wang, Jason Mills, Abhinav Diwan, and Deborah Frank for critical comments on the manuscript.
Funding
This work was funded in part by T32 HD049305 (to KAB) and R01 DK100644 (to IUM).
References
- Foxman B. The epidemiology of urinary tract infection. Nat Rev Urol 2010; 7:653-60; PMID:21139641; http://dx.doi.org/10.1038/nrurol.2010.190
- Flores-Mireles AL, Walker JN, Caparon M, Hultgren SJ. Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nat Rev Microbiol 2015; 13:269-84; PMID:25853778; http://dx.doi.org/10.1038/nrmicro3432
- Barber AE, Norton JP, Spivak AM, Mulvey MA. Urinary tract infections: current and emerging management strategies. Clin Infect Dis 2013; 57:719-24; PMID:23645845; http://dx.doi.org/10.1093/cid/cit284
- Wang C, Symington JW, Mysorekar IU. ATG16L1 and pathogenesis of urinary tract infections. Autophagy 2012; 8:1693-4; PMID:22874553; http://dx.doi.org/10.4161/auto.21600
- Wang C, Mendonsa GR, Symington JW, Zhang Q, Cadwell K, Virgin HW, Mysorekar IU. Atg16L1 deficiency confers protection from uropathogenic Escherichia coli infection in vivo. Proc Natl Acad Sci USA 2012; 109:11008-13; PMID:22715292; http://dx.doi.org/10.1073/pnas.1203952109
- Yep A, McQuade T, Kirchhoff P, Larsen M, Mobley HL. Inhibitors of TonB function identified by a high-throughput screen for inhibitors of iron acquisition in uropathogenic Escherichia coli CFT073. MBio 2014; 5:e01089-1013; PMID:24570372; http://dx.doi.org/10.1128/mBio.01089-13
- Garcia EC, Brumbaugh AR, Mobley HL. Redundancy and specificity of Escherichia coli iron acquisition systems during urinary tract infection. Infect Immun 2011; 79:1225-35; PMID:21220482; http://dx.doi.org/10.1128/IAI.01222-10
- Gao Q, Wang X, Xu H, Xu Y, Ling J, Zhang D, Gao S, Liu X. Roles of iron acquisition systems in virulence of extraintestinal pathogenic Escherichia coli: salmochelin and aerobactin contribute more to virulence than heme in a chicken infection model. BMC Microbiol 2012; 12:143; PMID:22817680; http://dx.doi.org/10.1186/1471-2180-12-143
- Subashchandrabose S, Mobley HL. Back to the metal age: battle for metals at the host-pathogen interface during urinary tract infection. Metallomics 2015; 7(6):935-42; PMID:25677827; http://dx.doi.org/10.1039/c4mt00329b
- Wiles TJ, Kulesus RR, Mulvey MA. Origins and virulence mechanisms of uropathogenic Escherichia coli. Exp Mol Pathol 2008; 85:11-19; PMID:18482721; http://dx.doi.org/10.1016/j.yexmp.2008.03.007
- Hagan EC, Mobley HL. Haem acquisition is facilitated by a novel receptor Hma and required by uropathogenic Escherichia coli for kidney infection. Mol Microbiol 2009; 71:79-91; PMID:19019144; http://dx.doi.org/10.1111/j.1365-2958.2008.06509.x
- Hagan EC, Lloyd AL, Rasko DA, Faerber GJ, Mobley HL. Escherichia coli global gene expression in urine from women with urinary tract infection. PLoS Pathog 2010; 6:e1001187; PMID:21085611; http://dx.doi.org/10.1371/journal.ppat.1001187
- Shields-Cutler RR, Crowley JR, Hung CS, Stapleton AE, Aldrich CC, Marschall J, Henderson JP. Human urinary composition controls Siderocalin's antibacterial activity. J Biol Chem 2015; 290(26):15949-60; PMID:25861985; http://dx.doi.org/10.1074/jbc.M115.645812
- Brumbaugh AR, Smith SN, Mobley HL. Immunization with the yersiniabactin receptor, FyuA, protects against pyelonephritis in a murine model of urinary tract infection. Infect Immun 2013; 81:3309-16; PMID:23798537; http://dx.doi.org/10.1128/IAI.00470-13
- Silva B, Faustino P. An overview of molecular basis of iron metabolism regulation and the associated pathologies. Biochim Biophys Acta 2015; 1852:1347-59; PMID:25843914; http://dx.doi.org/10.1016/j.bbadis.2015.03.011
- Richardson DR, Lane DJ, Becker EM, Huang ML, Whitnall M, Suryo Rahmanto Y, Sheftel AD, Ponka P. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc Natl Acad Sci USA 2010; 107:10775-82; PMID:20495089; http://dx.doi.org/10.1073/pnas.0912925107
- Lunova M, Goehring C, Kuscuoglu D, Mueller K, Chen Y, Walther P, Deschemin JC, Vaulont S, Haybaeck J, Lackner C, et al. Hepcidin knockout mice fed with iron-rich diet develop chronic liver injury and liver fibrosis due to lysosomal iron overload. J Hepatol 2014; 61:633-641; PMID:24816174; http://dx.doi.org/10.1016/j.jhep.2014.04.034
- Kurz T, Terman A, Gustafsson B, Brunk UT. Lysosomes in iron metabolism, ageing and apoptosis. Histochem Cell Biol 2008; 129:389-406; PMID:18259769; http://dx.doi.org/10.1007/s00418-008-0394-y
- Kurz T, Eaton JW, Brunk UT. The role of lysosomes in iron metabolism and recycling. Int J Biochem Cell Biol 2011; 43:1686-97; PMID:21907822; http://dx.doi.org/10.1016/j.biocel.2011.08.016
- De Domenico I, Vaughn MB, Li L, Bagley D, Musci G, Ward DM, Kaplan J. Ferroportin-mediated mobilization of ferritin iron precedes ferritin degradation by the proteasome. EMBO J 2006; 25:5396-404 PMID:17082767; http://dx.doi.org/10.1038/sj.emboj.7601409
- Bauckman KA, Haller E, Flores I, Nanjundan M. Iron modulates cell survival in a Ras- and MAPK-dependent manner in ovarian cells. Cell Death Dis 2013; 4:e592; PMID:23598404; http://dx.doi.org/10.1038/cddis.2013.87
- Bauckman K, Haller E, Taran N, Rockfield S, Ruiz-Rivera A, Nanjundan M. Iron alters cell survival in a mitochondria-dependent pathway in ovarian cancer cells. Biochem J 2015; 466:401-13; PMID:25697096; http://dx.doi.org/10.1042/BJ20140878
- Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014; 509:105-9; PMID:24695223; http://dx.doi.org/10.1038/nature13148
- Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, Triantafellow E, Menon S, Wang Z, Honda A, Pardee G, et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol 2014; 16:1069-79; PMID:25327288; http://dx.doi.org/10.1038/ncb3053
- Bauckman KA, Owusu-Boaitey N, Mysorekar IU. Selective autophagy: Xenophagy. Methods 2014; 75:120-7; PMID:25497060; http://dx.doi.org/10.1016/j.ymeth.2014.12.005
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/10.4161/auto.19496
- Oppenheimer SJ. Iron and infection: the clinical evidence. Acta Paediatr Scand Suppl 1989; 361:53-62; PMID:2485586
- Ala A, Schilsky ML. Inherited metabolic liver disease. Curr Opin Gastroenterol 2004; 20:198-207; PMID:15703644; http://dx.doi.org/10.1097/00001574-200405000-00004
- Straud S, Zubovych I, De Brabander JK, Roth MG. Inhibition of iron uptake is responsible for differential sensitivity to V-ATPase inhibitors in several cancer cell lines. PloS One 2010; 5:e11629; PMID:20661293; http://dx.doi.org/10.1371/journal.pone.0011629
- Dayani PN, Bishop MC, Black K, Zeltzer PM. Desferoxamine (DFO)–mediated iron chelation: rationale for a novel approach to therapy for brain cancer. J Neuro-Oncol 2004; 67:367-77; PMID:15164994; http://dx.doi.org/10.1023/B:NEON.0000024238.21349.37
- Allen GF, Toth R, James J, Ganley IG. Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep 2013; 14:1127-135; PMID:24176932; http://dx.doi.org/10.1038/embor.2013.168
- Mysorekar IU, Hultgren SJ. Mechanisms of uropathogenic Escherichia coli persistence and eradication from the urinary tract. Proc Natl Acad Sci USA 2006; 103:14170-5; PMID:16968784; http://dx.doi.org/10.1073/pnas.0602136103
- Knowles LM, Zewe J, Malik G, Parwani AV, Gingrich JR, Pilch J. CLT1 targets bladder cancer through integrin alpha5beta1 and CLIC3. Mol Cancer Res 2013; 11:194-203; PMID:23204394; http://dx.doi.org/10.1158/1541-7786.MCR-12-0300
- Terman A, Kurz T. Lysosomal iron, iron chelation, and cell death. Antioxid Redox Signal 2013; 18:888-98; PMID:22909065; http://dx.doi.org/10.1089/ars.2012.4885
- Morse D, Lin L, Choi AM, Ryter SW. Heme oxygenase-1, a critical arbitrator of cell death pathways in lung injury and disease. Free Radic Biol Med 2009; 47:1-12; PMID:19362144; http://dx.doi.org/10.1016/j.freeradbiomed.2009.04.007
- Khan MI, Mohammad A, Patil G, Naqvi SA, Chauhan LK, Ahmad I. Induction of ROS, mitochondrial damage and autophagy in lung epithelial cancer cells by iron oxide nanoparticles. Biomaterials 2012; 33:1477-1488; PMID:22098780; http://dx.doi.org/10.1016/j.biom-aterials.2011.10.080
- Chen G, Jing CH, Liu PP, Ruan D, Wang L. Induction of autophagic cell death in the rat brain caused by iron. Am J Med Sci 2013; 345:369-374; PMID:23187302; http://dx.doi.org/10.1097/MAJ.0b0-13e318271c031
- Tait SW, Ichim G, Green DR. Die another way–non-apoptotic mechanisms of cell death. J Cell Sci 2014; 127:2135-2144; PMID:24833670; http://dx.doi.org/10.1242/jcs.093575
- Symington JW, Wang C, Twentyman J, Owusu-Boaitey N, Schwendener R, Núñez G, Schilling JD, Mysorekar IU. ATG16L1 deficiency in macrophages drives clearance of uropathogenic E. coli in an IL-1beta-dependent manner. Mucosal Immunol 2015; 8(6):1388-99; PMID:25669147; http://dx.doi.org/10.1038/mi.2015.7
- Schaale K, Peters KM, Murthy AM, Fritzsche AK, Phan MD, Totsika M, Robertson AA, Nichols KB, Cooper MA, Stacey KJ, et al. Strain- and host species-specific inflammasome activation, IL-1beta release, and cell death in macrophages infected with uropathogenic Escherichia coli. Mucosal Immunol 2015; 9(1):124-36; PMID:25993444; http://dx.doi.org/10.1038/mi.2015.44
- Nagamatsu K, Hannan TJ, Guest RL, Kostakioti M, Hadjifrangiskou M, Binkley J, Dodson K, Raivio TL, Hultgren SJ. Dysregulation of Escherichia coli alpha-hemolysin expression alters the course of acute and persistent urinary tract infection. Proc Natl Acad Sci USA 2015; 112:E871-80; PMID:25675528; http://dx.doi.org/10.1073/pnas.1500374112
- Boyle KB, Randow F. The role of ‘eat-me’ signals and autophagy cargo receptors in innate immunity. Curr Opin Microbiol 2013; 16:339-48; PMID:23623150; http://dx.doi.org/10.1016/j.mib.2013.03.010
- Anderson CP, Shen M, Eisenstein RSm Lei bold EA. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim Biophys Acta 2012; 1823:1468-83; PMID:22610083; http://dx.doi.org/10.1016/j.bbamcr.2012.05.010
- Goodall M, Thorburn A. Identifying specific receptors for cargo-mediated autophagy. Cell Res 2014; 24:783-4; PMID:24797431; http://dx.doi.org/10.1038/cr.2014.56
- Zukor H, Song W, Liberman A, Mui J, Vali H, Fillebeen C, Pantopoulos K, Wu TD, Guerquin-Kern JL, Schipper HM. HO-1-mediated macroautophagy: a mechanism for unregulated iron deposition in aging and degenerating neural tissues. J Neurochem 2009; 109:776-91; PMID:19250338; http://dx.doi.org/10.1111/j.1471-4159.2009.06007.x
- Sakaida I, Kyle ME, Farber JL. Autophagic degradation of protein generates a pool of ferric iron required for the killing of cultured hepatocytes by an oxidative stress. Mol Pharmacol 1990; 37:435-42; PMID:2314391
- Pullarkat V, Meng Z, Donohue C, Yamamoto VN, Tomassetti S, Bhatia R, Krishnan A, Forman SJ, Synold TW. Iron chelators induce autophagic cell death in multiple myeloma cells. Leuk Res 2014; 38:988-96; PMID:24998390; http://dx.doi.org/10.1016/j.leukres.2014.06.005
- Chen CW, Chen TY, Tsai KL, Lin CL, Yokoyama KK, Lee WS, Chiueh CC, Hsu C. Inhibition of autophagy as a therapeutic strategy of iron-induced brain injury after hemorrhage. Autophagy 2012; 8:1510-20; PMID:22909970; http://dx.doi.org/10.4161/auto.21289
- Soundravally R, Agieshkumar B, Daisy M, Sherin J, Cleetus CC. Ferritin levels predict severe dengue. Infection 2015; 43:13-19; PMID:25354733; http://dx.doi.org/10.1007/s15010-014-0683-4
- Pandey R, Rodriguez GM. A ferritin mutant of Mycobacterium tuberculosis is highly susceptible to killing by antibiotics and is unable to establish a chronic infection in mice. Infect Immun 2012; 80:3650-9; PMID:22802345; http://dx.doi.org/10.1128/IAI.00229-12
- Vardhan H, Bhengraj AR, Jha R, Singh Mittal A. Chlamydia trachomatis alters iron-regulatory protein-1 binding capacity and modulates cellular iron homeostasis in HeLa-229 cells. J Biomed Biotechnol 2009; 2009:342032; PMID:19688112; http://dx.doi.org/10.1155/2009/342032
- Mittra B, Andrews NW. IRONy OF FATE: role of iron-mediated ROS in Leishmania differentiation. Trends Parasitol 2013; 29:489-96; PMID:23948431; http://dx.doi.org/10.1016/j.pt.2013.07.007
- do Nascimento PR, Martins DR, Monteiro GR, Queiroz PV, Freire-Neto FP, Queiroz JW, Morais Lima AL, Jeronimo SM. Association of pro-inflammatory cytokines and iron regulatory protein 2 (IRP2) with Leishmania burden in canine visceral leishmaniasis. PloS One 2013; 8:e73873; PMID:24146743; http://dx.doi.org/10.1371/journal.pone.0073873
- Mava Y, Ambe JP, Bello M, Watila I, Nottidge VA. Urinary tract infection in febrile children with sickle cell anaemia. West Afr J Med 2011; 30:268-72; PMID:22669831
- Christopher GW. Escherichia coli bacteremia, meningitis, and hemochromatosis. Arch Intern Med 1985; 145:1908; PMID:3899040; http://dx.doi.org/10.1001/archinte.1985.00360100178031
- Gordon S, Trinchieri G. Innate resistance and inflammation. Curr Opin Immunol 2009; 21:1-2; PMID:19223161; http://dx.doi.org/10.1016/j.coi.2009.02.001
- Biasiotto G, Di Lorenzo D, Archetti S, Zanella I. Iron and neurodegeneration: is ferritinophagy the link? Mol Neurobiol 2015; 1-33; PMID:26468157; http://dx.doi.org/10.1007/s12035-015-9473-y
- Kong XY, Nesset CK, Damme M, Løberg EM, Lübke T, Mæhlen J, Andersson KB, Lorenzo PI, Roos N, Thoresen GH, et al. Loss of lysosomal membrane protein NCU-G1 in mice results in spontaneous liver fibrosis with accumulation of lipofuscin and iron in Kupffer cells. Dis Model Mech 2014; 7:351-62; PMID:24487409; http://dx.doi.org/10.1242/dmm.014050
- Dikshit N, Bist P, Fenlon SN, Pulloor NK, Chua CE, Scidmore MA, Carlyon JA, Tang BL, Chen SL, Sukumaran B. Intracellular uropathogenic E. coli exploits host Rab35 for iron acquisition and survival within urinary bladder cells. PLoS Pathog 2015; 11:e1005083; PMID:26248231; http://dx.doi.org/10.1371/journal.ppat.1005083
- Puigserver, P. Part I Chapter 6: Signaling Transduction and Regulation of Cell Metabolism. (Hoffman R, Benz E, Silverstein L, Heslop H, Weitz J, Anastasi J, eds.) In Hematology: Basic Principles and Practices, 6th Edition. Philadelphia: Churchill Livingstone, 2012: 55–65.
- Matsumoto T. Urinary tract infections in the elderly. Curr Urol Rep 2001; 2:330-3; PMID:12084261; http://dx.doi.org/10.1007/s11934-001-0073-1
- Arshad M, Seed PC. Urinary tract infections in the infant. Clin Perinatol 2015; 42:17-28 vii; PMID:25677994; http://dx.doi.org/10.1016/j.clp.2014.10.003
- Collard KJ. Iron homeostasis in the neonate. Pediatrics 2009; 123:1208-16; PMID:19336381; http://dx.doi.org/10.1542/peds.2008-1047
- Pfrimer K, Micheletto RF, Marchini JS, Padovan GJ, Moriguti JC, Ferriolli E. Impact of aging on urinary excretion of iron and zinc. Nutr Metab Insights 2014; 7:47-50; PMID:24932105; http://dx.doi.org/10.4137/NMI.S12977
- Detweiler K, Mayers D, Fletcher SG. Bacteruria and Urinary Tract Infections in the Elderly. Urol Clin North Am 2015; 42:561-8; PMID:26475952; http://dx.doi.org/10.1016/j.ucl.2015.07.002
- Bishop BL, Duncan MJ, Song J, Li G, Zaas D, Abraham SN. Cyclic AMP-regulated exocytosis of Escherichia coli from infected bladder epithelial cells. Nat Med 2007; 13:625-30; PMID:17417648; http://dx.doi.org/10.1038/nm1572