
ABSTRACT
Xenophagy has been studied in epithelial cells infected with Salmonella enterica serovar Typhimurium (S. Typhimurium). Distinct autophagy receptors target this pathogen to degradation after interacting with ubiquitin on the surface of cytosolic bacteria, and the phagophore- and autophagosome-associated protein MAP1LC3/LC3. Glycans exposed in damaged phagosomal membranes and diacylglycerol accumulation in the phagosomal membrane also trigger S. Typhimurium xenophagy. How these responses control intraphagosomal and cytosolic bacteria remains poorly understood. Here, we examined S. Typhimurium interaction with autophagy in fibroblasts, in which the pathogen displays limited growth and does not escape into the cytosol. Live-cell imaging microscopy revealed that S. Typhimurium recruits late endosomal or lysosomal compartments that evolve into a membranous aggregate connected to the phagosome. Active dynamics and integrity of the phagosomal membrane are requisite to induce such aggregates. This membranous structure increases over time to become an aggresome that engages autophagy machinery at late infection times (> 6 h postentry). The newly formed autophagosome harbors LC3 and the autophagy receptor SQSTM1/p62 but is devoid of ubiquitin and the receptor CALCOCO2/NDP52. Live-cell imaging showed that this autophagosome captures and digests within the same vacuole the aggresome and some apposed intraphagosomal bacteria. Other phagosomes move away from the aggresome and avoid destruction. Thus, host endomembrane accumulation resulting from activity of intracellular S. Typhimurium stimulates a novel type of aggrephagy that acts independently of ubiquitin and CALCOCO2, and destroys only a few bacteria. Such selective degradation might allow the pathogen to reduce its progeny and, as a consequence, to establish persistent infections.
Introduction
Selective macroautophagy targets distinct objects such as damaged organelles (mitophagy, reticulophagy, pexophagy), protein aggregates (aggrephagy) or microbes and viruses (xenophagy).Citation1 In selective macroautophagy, double-membrane vesicles engulf cytosol portions containing these objects. This event is followed by fusion of the resulting autophagosomes with late endosomes and lysosomes.Citation1 The initiation of phagophore formation, promoted by processing of the autophagic protein MAP1LC3/LC3 and subsequent coupling to phosphatidylethanolamine (PE), is essential for autophagosome formation. In this state, LC3–PE (LC3-II) interacts with autophagy receptors bound to cargo to form a selective type of autophagosome. Among the distinct types of selective macroautophagy known, xenophagy constitutes an important host defense mechanism against intracellular bacterial pathogens.Citation2 Xenophagy controls infections caused by pathogens such as Shigella flexneri, Listeria monocytogenes, Legionella pneumophila, Mycobacterium tuberculosis and Salmonella enterica, among others.Citation2-4 As a result, many of these bacteria have developed strategies that counterattack and overcome this cellular defensive response. These include interference in lipid conjugation of the phagophore- and autophagosome-associated protein LC3,Citation5 and, avoidance of either autophagy receptor recruitment or ubiquitin deposition on the bacterial surface.Citation6-9 Besides its direct role in pathogen clearance, xenophagy modulates infection affecting inflammation, antigen presentation, and secretion of immune mediators.Citation2,10
S. enterica serovar Typhimurium (S. Typhimurium) is an intracellular bacterial pathogen that infects phagocytic and nonphagocytic eukaryotic cells and resides in a specialized phagosomal compartment named the SCV, for Salmonella-containing vacuole.Citation11-13 In vivo, S. Typhimurium preferentially colonizes macrophages, both in target organs and in lymph nodes.Citation14 Once inside the macrophage, the pathogen shows limited proliferative capacity.Citation15 S. Typhimurium displays similar limited growth capacity in cultured fibroblastsCitation16,17 and in vivo inside nonphagocytic stromal cells located in the intestinal lamina propria.Citation18 Conversely, S. Typhimurium proliferates actively following entry into cultured epithelial cells.Citation19 Most reports using cultured epithelial cell lines describe net increases in intracellular bacterial loads up to 30- to 40-fold.Citation19,20 Active growth of this pathogen inside epithelial cells has attracted most studies involving S. Typhimurium adaptation to an intracellular lifestyle. S. Typhimurium proliferation within epithelial cells associates to a profound reorganization of host endomembranes.Citation11 The pathogen subverts vesicular trafficking by targeting motor proteins such as dynein and kinesin, which leads to formation of tubules termed SIF, for Salmonella-induced filaments.Citation21 SIF are enriched in lysosomal membrane glycoproteins (LGP), emanate from the SCV and are proposed as the main source of membrane and nutrients for proliferation of intraphagosomal bacteria.Citation21,22 Besides SIF, other tubular structures are reported in S. Typhimurium-infected epithelial cells.Citation22 How these distinct tubular networks modulate the intracellular lifestyle of the pathogen is currently unknown.
In epithelial cells, xenophagy targets S. Typhimurium following recognition of bacteria that escape from the SCV into the cytosol or membranes derived from damaged SCV.Citation23 Pathogen recognition occurs by ubiquitination of surface components in cytosolic bacteriaCitation24-27 or via interaction with LGALS8/galectin-8, a cytosolic lectin that binds glycans exposed on the luminal side of damaged SCV membranes.Citation28 Ubiquitinated components on the pathogen surface and LGALS8 act as danger sensors recognized by diverse autophagy receptors, including CALCOCO2/NDP52, SQSTM1/p62, OPTN/optineurin and CALCOCO3/TAX1BP1.Citation23,29-31 Diacylglycerol (DAG) signaling can also target S. Typhimurium to xenophagy.Citation32 These xenophagy events occur at early stages of the infection, 1 to 2 h after bacterial invasion of the epithelial cell. The mode in which this early xenophagy phase connects to the subsequent period of active proliferation of the pathogen (starting at 4 to 6 h postinfection) has not been yet analyzed. A recent report provided evidence for a role of autophagy in repairing some damaged phagosomes harboring S. Typhimurium.Citation33 Another study, however, showed that S. Typhimurium subverts autophagy to proliferate in the cytosol of epithelial cells.Citation19 The precise contribution of autophagy to the balance of the intraphagosomal and cytosolic bacterial populations coexisting in the epithelial cell remains not completely understood.
Unlike epithelial cells, in which up to 70% of S. Typhimurium-infected cells show SIFs associated with the SCV,Citation34 fibroblasts harboring nongrowing persistent S. Typhimurium rarely form SIF and bacteria do not escape into the cytosol.Citation17 These differences manifest that SCV biogenesis follows distinct routes in these 2 nonphagocytic cell types. A more restrictive SCV environment could contribute to the nonproliferative state that S. Typhimurium establishes inside fibroblasts. Considering the contrasting roles assigned to xenophagy in restriction or promotion of S. Typhimurium proliferation in epithelial cells,Citation19,26,35 we determined whether xenophagy contributes to the nonproliferative lifestyle that S. Typhimurium adopts in fibroblasts. Live-cell imaging microscopy uncovered a novel aggrephagy event involving digestion of an aggresome composed of host cell endomembranes and generated as a result of the infection. This digestion also affects some SCV located close to the aggregate. The implications of these findings are discussed in a model involving a novel type of autophagy manipulation exploited by the pathogen to reduce its progeny inside the infected cell.
Results
S. typhimurium induces in fibroblasts aggregates enriched in lysosomal membrane glycoproteins
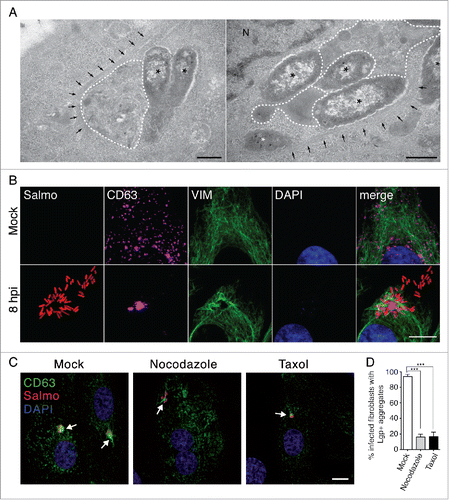
Following entry into cultured fibroblasts, S. Typhimurium progeny remains fairly constant (1- to 5-fold increases in bacterial numbers) for long periods of time.Citation16,17 We hypothesized that a distinct SCV biogenesis could be responsible for this nonproliferative state. As SCV accumulate lysosomal membrane glycoproteins (LGP) in epithelial cells,Citation22,34,36 we examined the distribution of SCARB2/Limp-II, a lysosomal integral membrane protein in S. Typhimurium-infected NRK-49F fibroblasts, fixed and processed for immunofluorescence microscopy. For comparison, we also monitored distribution of another LGP, LAMP1 (lysosomal-associated membrane protein 1) in infected HeLa epithelial cells. In agreement with previous studies,Citation34,36 the infected epithelial cell showed long SIF connected to LAMP1-positive SCV (). These structures were visualized at 8 to 16 h postinfection (hpi) but were lost later in the infection, by 24 h (). In contrast, the SCV in fibroblasts were apposed to SCARB2-positive “aggregate”-like structures at 2 to 8 hpi (). At later times, 16 hpi, we observed in the infected fibroblast large vacuoles harboring both the aggregate and some SCV (). The strong fluorescence signal associated with the aggregate made it difficult to visualize the presence of LGP in the phagosomal membrane when short exposures were used for image acquisition. Despite this, intracellular bacteria were visible inside LGP-positive phagosomes at the stage of “early” aggregates, 8 hpi (inset, ) and also at later infection times, 16 hpi, when the aggregate evolved into a much larger structure (inset, ). At 24 hpi, most intracellular bacteria were inside individual phagosomes displaying a scattered distribution (inset, ). In contrast to epithelial cells,Citation37 we did not detect intracellular bacteria devoid of the LGP markers at any postinfection time. This observation confirmed that S. Typhimurium does not escape into the cytosol of fibroblasts. LGP+ aggregates also formed near SCV in fibroblasts of other origin (human) and containing other LGP such as LAMP1/CD107a, LAMP2/CD107b and CD63 (Fig. S1). Of note, the CD63 protein, also known with the alias LAMP-3 in some databases, is a lysosomal membrane glycoprotein of 238 amino acids unrelated to the LAMP3/CD208 protein of 416 amino acids. For clarification purposes, we will refer to this LGP hereinafter exclusively as CD63.
Figure 1. Distinct biogenesis of Salmonella-containing vacuole (SCV) in fibroblasts and epithelial cells. (A) Confocal microscopy images of the SCV at the indicated postinfection times in HeLa human epithelial and NRK-49F rat fibroblasts. LAMP1 and SCARB2 lysosomal membrane glycoproteins (LGP; green) were detected as late endosome/lysosome markers in HeLa and NRK-49F cells, respectively. In infected NRK-49F fibroblasts, arrows indicate an LGP+ aggregate (8 hpi), a large vacuole (16 hpi), and an isolated/individual SCV (24 hpi) that are enlarged in the respective insets; scale bar: 10 μm. (B) Percentage of infected fibroblasts containing LGP+ aggregates and large vacuoles quantified by time-lapse microscopy at the indicated postinfection times. Stably transfected NRK-49F fibroblasts expressing CD63GFP (green) were used for this experiment. Data were obtained from 3 independent experiments in which at least 100 cells were examined per assay. (C) Time-lapse microscopy frames showing distinct steps of SCV biogenesis in fibroblasts that are not seen in epithelial cells; LGP+ aggregate (Agg) and large vacuole (LV) indicated at 11 hpi; scale bar: 5 μm. (D) Time-lapse series showing a peduncle-like connection between an LGP+ aggregate and 2 SCV. Images were acquired over a 10-min period starting at 4 hpi (time 0); scale bar: 5 μm. hpi, hours postinfection.
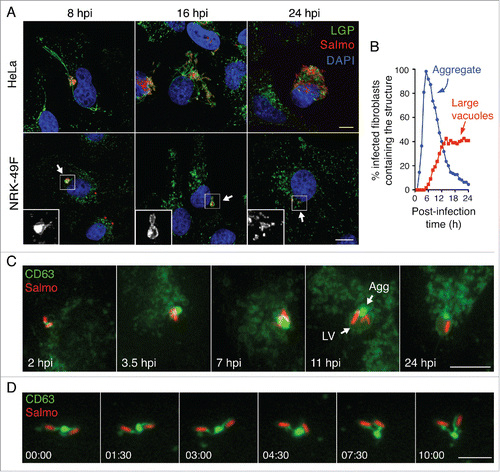
The transient nature of the LGP+ aggregate in the infected fibroblast followed by the appearance of large vacuoles suggested a probable functional relationship between the 2 structures. To test this, we generated stably transfected NRK-49F fibroblasts expressing CD63-GFP. More than 100 cells per experiment were tracked by live-cell imaging microscopy over a period of 24 h. This analysis revealed LGP+ aggregates in most infected fibroblasts, with a maximum of ∼95% of this population harboring the aggregate at 5 hpi (). Live-cell imaging also showed that these LGP+ aggregates increase in size over time to be subsequently engulfed by large vacuoles. Other phenomena visualized were intimately associated bacteria inside the large vacuoles that harbored the LGP+ aggregate and the gradual fading of the material (aggregate and bacteria) enclosed within this compartment (). Time-lapse and confocal microscopy showed that the engulfed bacteria were previously enclosed in individual phagosomes () and that SCV and LGP+ aggregates were connected by peduncle-like structures (). These connections were visible during early infection stages when LGP+ aggregates were small enough for not masking such structure (). The presence of LGP in all SCV examined was consistent with a homogeneous population of intracellular bacteria residing within phagosomes. Taken together, these data showed that the formation of LGP+ aggregates is a distinctive feature of S. Typhimurium infections progressing with limited pathogen proliferation.
The LGP+ aggregate induced by S. typhimurium in fibroblasts shares SIF features
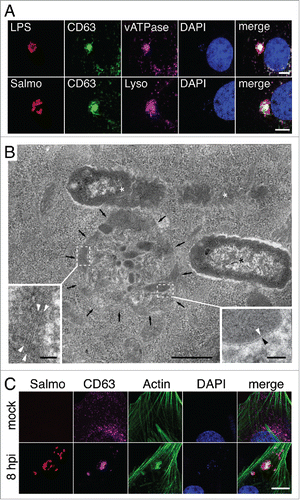
Live-cell imaging microscopy allowed us to occasionally observe SIF-like filaments in S. Typhimurium-infected fibroblasts. These SIF-like filaments exhibited the elongation, retraction and branching reported for SIF in epithelial cellsCitation38,39 (Fig. S2, video S6). However, unlike the SIF reported in epithelial cells, the SIF-like filaments observed in fibroblasts were transient and did not evolve to a stable network (Fig. S2, video S6). This difference might explain why SIF are rarely observed in fixed fibroblast cells (). Another distinctive feature of the SIF-like filaments in fibroblasts was that they emerge from the LGP+ aggregate induced by S. Typhimurium (video S6). This observation led us to characterize the composition and ultrastructure of the aggregate. The LGP+ aggregate accumulates LysoTracker Deep Red and has vacuolar ATPase (v-ATPase) (), 2 markers of acidic compartments also present in SIF formed in infected epithelial cells. Correlative light and electron microscopy (CLEM) showed that the LGP+ aggregate consisted of a cluster of “membrane-bound compartments” showing vesicular or tubular shape depending on the section angle (, S3, S4). Many of the structures appearing as small vesicles in the transmission electron microscopy (TEM) analysis were tracked for several serial sections of ∼50 to 70 nm, denoting that they are tubules (Fig. S4A and B). Regular fibers resembling cytoskeletal elements were also seen inside these membranous compartments (). Confocal microscopy confirmed the presence of polymerized actin in the LGP+ aggregate (), a feature reported for the SIF formed in epithelial cells infected with S. Typhimurium.Citation40 The LGP+ aggregate is therefore compatible with membranous SIF-like tubules packed like a “crumpled” paper ball. Altogether, these data suggest that, despite their different morphologies, the LGP+ aggregates in fibroblasts and the SIF in infected epithelial cells have a common origin.
Figure 2. The LGP+ aggregates induced by S. Typhimurium in fibroblasts share characteristics with SIF. (A) Confocal microscopy sections showing colocalization of LGP+ aggregates (green) with vacuolar (v)ATPase and LysoTracker Deep Red (magenta). Bacteria were identified with anti-S. Typhimurium lipopolysaccharide (LPS) antibody or using a strain expressing DsRed (Salmo, red). Scale bar: 5 μm. (B) Electron microscopy of an LGP+ aggregate (black arrows). Insets show the membranous nature of the tubulo-vesicular compartments (right; arrowheads indicate a lipid bilayer) and fibrillar structures inside a membranous compartment (left; arrowheads indicate electron-dense filaments). Asterisks, intracellular bacteria enclosed within SCV. Scale bars: 500 nm (central image), 50 nm (insets). (C) Confocal microscopy showing the presence of polymerized actin in the LGP+-aggregate. Images were obtained at 8 hpi from NRK-49F stably transfected with CD63-GFP and infected with DsRed-expressing S. Typhimurium. Bacteria: Salmo, red; CD63: magenta; actin: green; DAPI: blue. Scale bar: 5 μm.
The LGP+ aggregate is digested by ubiquitin-independent aggrephagy that also affects phagosomes located in the vicinity
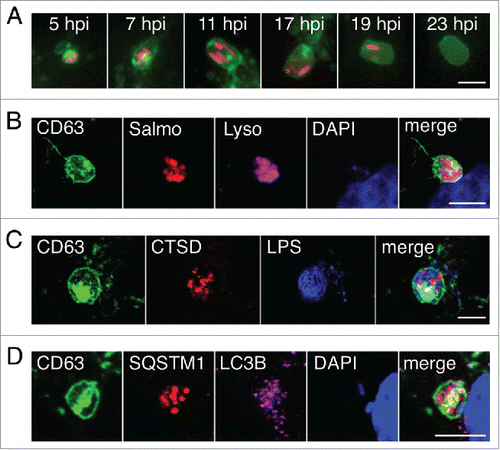
Fibroblasts infected persistently with S. Typhimurium are viable for up to 3 wk.Citation41 We hypothesized that an effective response to the “aberrant” membranous aggregate could be essential to ensure coexistence of the pathogen and the infected fibroblast. Live-cell imaging showed that the infected fibroblast digests the LGP+ aggregates within large vacuoles, a process that lasts up to 10 to 12 h (). Fluorescence intensity derived from bacteria and/or LGP+ aggregates was stable over time only if positioned outside the large vacuoles. This observation suggested that the material inside the large vacuoles disappeared as a consequence of active degradation, i.e. an autophagy event. To test this, we examined the presence of different autophagy markers in the large vacuoles. Unlike the SCV, the large vacuoles contained CTSD (cathepsin D), the phagophore and autophagosome protein LC3 (the LC3B isoform is recognized by our antibody) and the autophagy receptor SQSTM1 (). These data demonstrated that the large vacuoles have an autophagy origin and are degradative compartments responding to the LGP+ aggregate. This assumption was supported by the marked decreased in intracellular bacterial load when the infected fibroblasts were subjected to starvation, known to stimulate autophagy,Citation42 at the time that the LGP+ aggregates were already formed (Fig. S5).
Figure 3. The large vacuoles that engulf the LGP+ membranous aggregates are autophagosomes. (A) Live-cell imaging microscopy of a stably transfected NRK-49F fibroblast expressing CD63-GFP, showing an LGP+ aggregate (green) and 2 SCV (red) that are engulfed simultaneously and digested within a large vacuole. (B-D), Confocal microscopy images of stably transfected CD63-GFP NRK-49F fibroblasts showing: (B) large vacuoles with acidic pH (LysoTracker Deep Red-positive; Lyso; magenta); (C) large vacuoles containing CTSD (red); and, (D) large vacuoles positive for the autophagosome markers SQSTM1 (red) and LC3B (magenta). Images were acquired at 16 hpi. Bacteria are identified by DsRed expression (B); recognition of S. Typhimurium LPS (blue) (C); or DAPI (blue) (D). Scale bars: 5 μm.
Live-cell imaging microscopy showed concomitant degradation of the LGP+ aggregates and intimately associated bacteria that shared the same large vacuole (). Macroautophagy provides the most efficient pathway to clear aggresomes, a phenomenon generally known as aggrephagy.Citation43 Although described as essential for aggrephagy,Citation43,44 we did not detect ubiquitin recruitment to the LGP+ aggregates or to the nearby SCV at 8 hpi (), a time at which the autophagosome initiates its formation (). Absence of ubiquitin in the LGP+ aggregate at 8 hpi was confirmed after examining in 3 independent experiments more than 500 infected fibroblasts at this infection time. None of these cells showed specific ubiquitin label in the aggregate. Lack of ubiquitin around the LGP+-aggregate was also evident when overexposing the immunofluorescence signal to visualize ubiquitin distribution in the entire cell ().- CALCOCO2, an autophagy receptor that targets cytosolic bacteria and damaged or broken vacuoles,Citation28,29 was also absent in the LGP+ aggregates and nearby SCV at the same postinfection time, 8 h (). These observations indicated that the aggrephagy of LGP+ aggregates induced by S. Typhimurium occurs in a ubiquitin-independent manner. In addition, the absence of CALCOCO2 in the LGP+ aggregate suggested that the compartments recruited during aggresome formation preserve their membrane integrity.
Figure 4. Selective autophagy of LGP+ membranous aggregates differs from Salmonella xenophagy. (A to C) Confocal microscopy images showing that ubiquitin (A, B) and CALCOCO2 (C) decorate damaged Salmonella-containing vacuoles (SCV) at 2 hpi but are not detected at 8 hpi in the LGP+ membranous aggregates. Panel B, in which the fluorescence signal was processed to appear with higher intensity, shows ubiquitinated components in the entire cell. Arrows indicate intracellular bacteria. Ubiquitin was detected in NRK49F rat fibroblasts whereas CALCOCO2 was detected in BJ-5ta human fibroblasts. Bacteria (dsRed); Ubiquitin (UBIQ). Scale bars: 5 μm (A, (C)panels), 10 μm (B panel). (D) Percentage of ubiquitin- and CALCOCO2-positive SCV in fibroblasts and epithelial cells quantified at 2 hpi by epifluorescence microscopy. Data are the means and standard deviations from 3 independent experiments. n.s., not significant (Student t test).
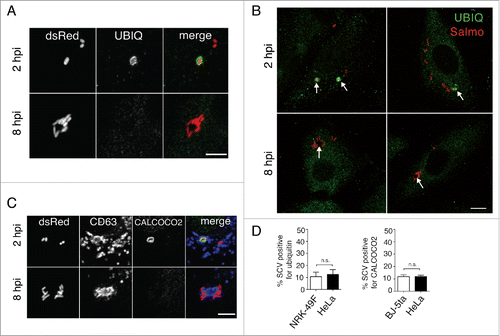
Of interest, when we examined the distribution of CALCOCO2 and ubiquitin in the fibroblast at earlier infection times, 2 hpi, ∼10% of SCV were positive for these markers (). Presence of these markers was detected exclusively at those early infection times, 1 to 2 hpi (), preceding the formation of the LGP+ aggregates that initiates at 4 hpi (). We found a similar percentage of SCV positive for CALCOCO2 and ubiquitin in S. Typhimurium-infected HeLa epithelial cells (), which agrees with previous reports.Citation24,45,46 Altogether, these data suggest that in fibroblasts, as in epithelial cells, there is a first autophagy event directed against S. Typhimurium that can be classified as xenophagy. This xenophagy, however, occurs independently and differs in terms of markers, timing, and autophagosome dimensions from the second autophagy wave (aggrephagy) that is exclusive to fibroblasts and initiates at 6 hpi ().
The LGP+ aggregate is surrounded by VIM and directed to the MTOC by microtubules
Aggresome formation is a general cellular response to the presence of aggregated proteins exceeding the proteolysis potential of the cell. Aggresomes are pericentriolar cytoplasmic protein inclusions that are ubiquitinated and sheathed in a cage of intermediate filaments.Citation47 Aggresome formation initiates by aggregation of misfolded proteins into small bodies that subsequently traffic toward the microtubule-organizing center (MTOC) in a microtubule-dependent manner. Fusion of these small bodies results in a single large aggresome.Citation48,49 Macroautophagy targets these aggresomes for digestion.Citation43,50 Classical aggresomes differ from other protein aggregates formed under a wide range of stress conditions such as aggresome-like induced structures (ALIS)Citation51 or dendritic cell aggresome-like induced structures (DALIS).Citation52 Although ALIS and DALIS resemble aggresomes morphologically, these 2 structures are formed independently of microtubules and do not colocalize with the MTOC. Moreover, ALIS and DALIS are not trapped by VIM (vimentin) cages.Citation51,52
Ultrastructural TEM analysis of the tubule-vesicular membranous LGP+ aggregate formed in the S. Typhimurium-infected fibroblast showed fibers that resembled intermediate filaments (). Confocal microscopy supported the presence of VIM filaments decorating the LGP+ aggregate (). Image stacks along the Z-axis unequivocally showed that VIM cages trapped the LGP+ aggregate and some of the SCV located in the vicinity (Fig. S6, video S7). These data demonstrated that the LGP+ aggregates are bona fide aggresomes segregated from other cell contents by VIM.Citation53 Disruption of microtubule (MT) dynamics with drugs as nocodazole or taxol resulted in loss of most of the LGP+ aggregates (). Only ∼16% of infected cells showed clear LGP+ aggregates in the drug-treated fibroblasts versus ∼94% of infected cells with such aggregates in the untreated fibroblasts (). MT dynamics is therefore essential for the formation of the LGP+ aggregate induced by S. Typhimurium. Positioning of the LGP+ aggregate in the MTOC area of the infected fibroblast () agreed with this requirement. Taken together, these data show that the LGP+ aggregate induced by S. Typhimurium in fibroblasts is a classical aggresome distinct from previously described ALIS or DALIS structures.
Figure 5. The LGP+ membranous aggregate is segregated by VIM cages and depends on microtubule dynamics. (A) TEM micrographs showing LGP+ aggregates (dotted lines) surrounded by fibril bundles compatible with VIM intermediate filaments; asterisks, intracellular bacteria; black arrows, fibril bundles compatible with VIM. Scale bar: 500 nm. (B) Confocal microscopy sections showing a VIM cage surrounding an LGP+ aggregate and some intracellular bacteria. CD63: magenta; bacteria: Salmo, red; VIM: green; DAPI: blue. Scale bar: 5 μm. (C) Confocal microscopy Z-projections showing morphology of the LGP+ aggregates in fibroblasts pretreated with 10 μg/ml nocodazole, 10 μM taxol or mock treated (see Methods for details). CD63: green; bacteria: Salmo, red; DAPI: blue. Scale bar: 10 μm. Images were obtained in all assays at 8 hpi from NRK-49F stably transfected with CD63-GFP and infected with DsRed-expressing S. Typhimurium. (D) Effect of drugs disrupting MT dynamics in formation of the LGP+ aggregate. NRK-49F-CD63-GFP fibroblasts were pretreated with 10 μg/ml nocodazole or 10 μM taxol, infected, fixed at 8 hpi and examined by epifluorescence microscopy for the presence of the LGP+ aggregate. Data are the means and standard deviations from 3 independent experiments. ***, P < 0.001 (Student t test).
The LGP+ aggregate is a highly insoluble aggresome
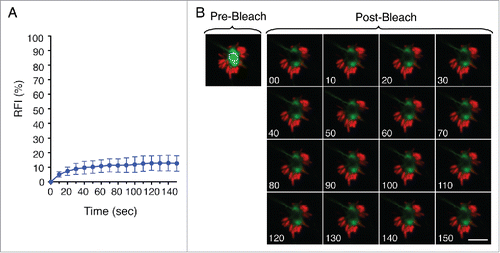
A recent study proposed a novel recognition mechanism by macroautophagy based on aggregate dynamic properties rather than the nature of the aggregated material.Citation54 This study analyzed clearance of SNCAIP/synphilin-1 aggregates, often found as pathogenic protein inclusions.Citation54 Such observation led us to examine the dynamic properties of the LGP+ aggregate induced by S. Typhimurium by fluorescence recovery after photobleaching (FRAP) analysis. FRAP of late LGP+ aggregates (8 hpi) showed ∼10% fluorescence recovery, consistent with a highly static and insoluble structure (). Apart from the membranous nature of the aggresome that contributes to constrain mobility, other forces might facilitate its progressive compaction. These data indicated that the aggrephagy event that takes place in the S. Typhimurium-infected fibroblast could be responding to the physical properties of a highly insoluble and static LGP+ aggregate.
Figure 6. The LGP+ membranous aggregate is a static structure. (A) Recovery of fluorescence intensity (RFI) after photobleaching of LGP+ aggregates. Fluorescence recovery measurement began at 8 hpi and bleaching focused on central region of aggregates. Data show mean ± SD of 10 LGP+ aggregates from 2 independent experiments. (B) Time-lapse series of a representative LGP+ aggregate before (pre-) and after (post-) photobleaching. The analysis was done with stably transfected NRK49F fibroblasts expressing CD63-GFP (green) infected with DsRed-S. Typhimurium (red). Scale bar: 5 μm.
The type III secretion system encoded in SPI2 is required to induce the LGP+-aggregate
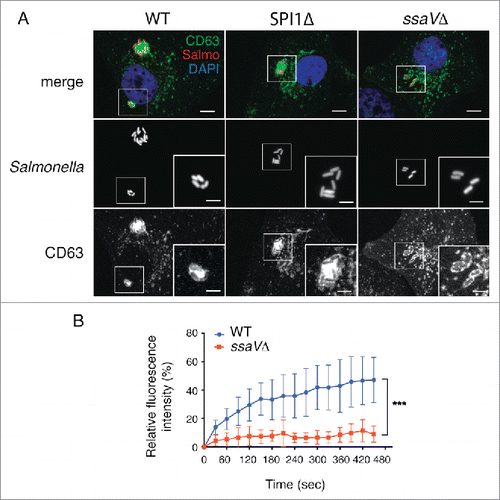
So far, our data support a model of aggresome formation in fibroblasts based on alteration of SIF dynamics shortly after their formation. In epithelial cells, SIF formation is promoted by effector proteins that translocate via the type III secretion system encoded in Salmonella pathogenicity island 2 (SPI2-T3SS).Citation21,39,55 Infection of fibroblasts with a S. Typhimurium mutant defective for secretion of SPI2 effectors (ssaVΔ) revealed that SPI2-T3SS is necessary for formation of the LGP+ aggregate (). This negative result was confirmed after visualizing in 3 independent experiments more than 300 individual cells infected with this ssaVΔ mutant. Conversely, a SPI1Δ mutant lacking the invasion-associated SPI1-T3SS induced aggresomes indistinguishable from those induced by wild-type bacteria (). FRAP experiments confirmed that SPI2-T3SS is needed for recruitment of membranous material to the SCV (). Incorporation of new membrane material, which depends on active dynamics in the phagosomal membrane, might be therefore essential to induce the LGP+ aggregate. FRAP assays showed that SCV containing wild-type bacteria recovered ∼40% of CD63-GFP fluorescence 5 to 6 min postphotobleaching whereas phagosomes harboring the ssaVΔ mutant recovered <10% of the initial fluorescence intensity (). These data supported the idea that, at an early infection stage in fibroblasts, SPI2 protein effectors provide the required dynamics to the phagosomal membrane to initiate formation of the SIF-like filaments.
Figure 7. The Salmonella pathogenicity island 2 (SPI2) is required for induction of the LGP+ aggregate in fibroblasts. (A) Confocal microscopy images showing stably transfected NRK49F fibroblasts expressing CD63-GFP (green) infected with S. Typhimurium wild-type (WT) or SPI1Δ strains expressing DsRed (red). No LGP+ aggregate was detected in fibroblasts infected with an isogenic ssaVΔ mutant expressing DsRed. Images obtained at 8 hpi. Insets show groups of bacteria with the LGP+ aggregate. Scale bars: 5 μm (original images), 2.5 μm (insets). (B) FRAP analysis of SCV containing WT and ssaVΔ bacteria. Fluorescence recovery was measured starting at 3 hpi. Data shown as means ± standard deviations of SCV from 2 independent experiments (n = 10). ***, P < 0.001 (Student t test).
The SPI2 effectors SseL and SteA are not involved in the induction of the LGP+ aggregate
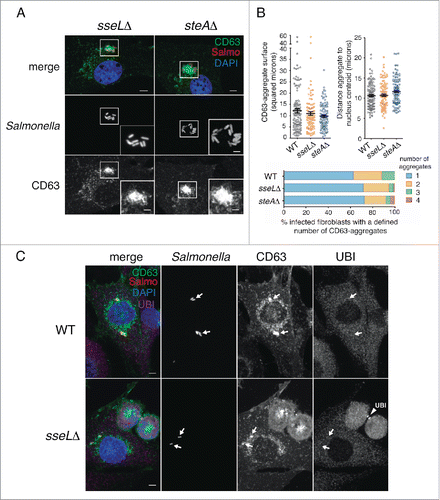
The results obtained with the ssaVΔ mutant () implicated effectors translocated by SPI2-T3SS in the formation of the LGP+ aggregate responsible for the subsequent aggrephagy event. To assess the role of defined SPI2 effectors, we first tested SseL, a SPI2 effector with deubiquitinase activity.Citation9 In epithelial cells, SseL modulates negatively the autophagy response to S. Typhimurium.Citation9 Fibroblasts infected with the sseLΔ mutant showed LGP+ aggregates similar in size, subcellular location and number per cell to those induced by the wild-type bacteria (). These LGP+ aggregates were also devoid of ubiquitin (), which discarded a major role of SseL in the aggrephagy event. These data provided further support to the ubiquitin-independent recognition of the LGP+ aggregate. We next examined the SPI2 effector SteA. In epithelial cells, lack of SteA in the infecting bacteria results in LAMP1 accumulation near the SCV and formation of large vacuoles and aggregates that were cataloged as ALIS.Citation56 Fibroblasts infected with the steAΔ mutant exhibited LGP+ aggregates similar to those induced by the wild-type bacteria (). Live-cell imaging microscopy in CD63-GFP expressing HeLa epithelial cells infected with the steAΔ mutant showed LGP+ aggregates that were digested within large vacuoles harboring autophagosome markers (Fig. S7), which suggests that aggrephagy could also occur in epithelial cells when bacteria lacks defined SPI2 effectors. In this HeLa cell infection model, the LAMP1 aggregates and the abnormal vacuoles were previously reported as separate entities.Citation56 Of interest, wild-type bacteria translocate SseL and SteA inside the fibroblast, as revealed by western blot (Fig. S8). Altogether, these data demonstrated that SseL and SteA are dispensable for the formation of the LGP+ aggregate and the subsequent aggrephagy that are observed in infected fibroblasts.
Figure 8. The SPI2 effectors SseL and SteA do not contribute to formation of the LGP+ aggregate. (A) Confocal microscopy images showing LGP+ aggregates in CD63-GFP expressing NRK-49F fibroblasts infected with sseLΔ and steAΔ mutant strains expressing DsRed. Images obtained at 8 hpi. Insets show groups of bacteria with their associated LGP+ aggregates. Bacteria: Salmo, red; CD63: green; DAPI: blue. Scale bars: 5 μm (original images), 2.5 μm (insets). (B) Quantitative data of parameters such as: surface of the LGP+ (CD63)-aggregate, distance between the LGP+ aggregate and the nucleus centroid, and number of LGP+ aggregates per cell. At least 100 LGP+ aggregates or fibroblasts containing LGP+ aggregates were counted in 2 independent experiments. (C) The lack of SseL does not result in ubiquitin accumulation around the SCV or the LGP+ aggregate. CD63-GFP expressing NRK-49F fibroblasts were infected with the sseLΔ mutant expressing DsRed. Cell were fixed at 8 hpi and processed for immunofluorescence microscopy using anti-ubiquitin (UBI) antibody. Bacteria: Salmo, red; CD63: green; Ubiquitin: UBI, magenta; DAPI: blue. Scale bars: 5 μm. Arrows indicate positioning of bacteria and LGP+ aggregates. Note as positive control for the anti-ubiquitin antibody the labeling of ubiquitin in the midbody of 2 dividing cells in last stages of cytokinesis (arrowhead, UBI).
A combined action of the SPI2 effectors SifA, SseJ, and SopD2 ensures formation of large LGP+ aggregates positioned in the MTOC
The LGP+ aggregate is formed concomitantly to the loss of movement in the transient SIF-like structures visualized early in the infection (Fig. S2). As SifA is a SPI2 effector required for SIF formation,Citation57 we investigated its role in fibroblasts. In the epithelial cell infection model, the lack of SifA results in instability of the SCV membrane and increased number of hyper-replicating cytosolic bacteria.Citation57 Of interest, the simultaneous loss of SifA and other SPI2 effectors such as SseJ or SopD2, leads to increased stability of the phagosomal membrane.Citation58,59 Infection of fibroblasts with sifAΔ, sseJΔ and sopD2Δ single mutants showed that SifA is required for formation of the LGP+ aggregate (). Noteworthy, the double mutants sifAΔ sseJΔ and sifAΔ sopD2Δ regained capacity to induce the formation of LGP+ aggregates (). Loss of either SseJ or SopD2 led to a higher average number of LGP+ aggregates per cell (), although of smaller size (). These defects were more pronounced for the LGP+ aggregates induced by the sifAΔ sseJΔ and sifAΔ sopD2Δ double mutants, which were visualized away from the MTOC (). The phenotypes shown by these single and double mutants remained unaltered at later post infection times, 16 hpi (Fig. S9).
Figure 9. The SPI2 effector SifA is required for LGP+ aggregate formation with a combined action of SseJ and SopD2. (A) Confocal microscopy images showing CD63-GFP expressing NRK-49F fibroblasts infected with wild-type (WT), and sifAΔ, sseJΔ, and sopD2Δ single mutants expressing DsRed. Images obtained at 8 hpi. Insets show groups of bacteria with their associated LGP+ aggregates (arrows), except in the case of the sifAΔ, in which are not formed. Bacteria: Salmo, red; CD63: green; DAPI: blue. Scale bars: 5 μm. (B) Confocal microscopy images showing CD63-GFP expressing NRK-49F fibroblasts infected with sifAΔ sseJΔ and sifAΔ sopD2Δ double mutants expressing DsRed. Images obtained at 8 hpi. Insets show groups of bacteria with their associated LGP+ aggregates (arrows). Bacteria: Salmo, red; CD63: green; DAPI: blue. Scale bars: 5 μm. (C) Quantification of surface of the LGP+ (CD63)-aggregate and distance between the LGP+ aggregate and the nucleus centroid in CD63-GFP expressing NRK-49F fibroblasts infected with the indicated isogenic strains. At least 100 LGP+ aggregates were counted in 2 independent experiments. Data obtained at 8 hpi. **, P<0.01; ***, P <0.001 (Kruskal-Wallis test with the Dunn post-hoc test). (D) Quantification of number of LGP+ aggregates visualized per fibroblast infected with the indicated isogenic strains. At least one hundred LGP+ aggregates were examined in 2 independent experiments. Data obtained at 8 hpi. (E) Scheme denoting the capacity of the distinct isogenic strains used to induce the formation of large LGP+ aggregates. Relative size of the LGP+ aggregate is shown proportional to the average surface measured for each strain (see panel C).
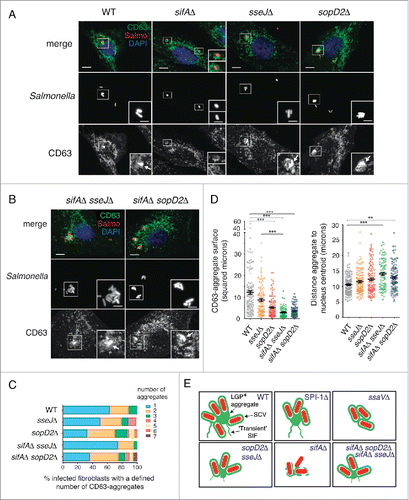
Together, these observations indicated that besides an active dynamics in the phagosomal membrane, its integrity is also a requisite for formation of the LGP+ aggregate (). Thus, whereas the sifAΔ mutant does not induce LGP+ aggregates (), the sifAΔ sseJΔ and sifAΔ sopD2Δ double mutants enclosed in intact phagosomes are capable of inducing such membranous aggregates (). Membrane integrity in mature phagosomes must be therefore an inherent feature of the SCV in fibroblasts, which might explain why LGP+ aggregates are not formed in epithelial cells. This statement is supported by the fact that < 5% of intracellular S. Typhimurium reach the cytosol in fibroblasts while ∼50% cytosolic bacteria are counted in epithelial cells at 8 hpi, as shown by digitonin-based permeabilization assays (Fig. S10). Thus, unlike SseL and SteA, other SPI2 effectors such as SifA, SseJ, and SopD2 contribute to the formation of large LGP+ aggregates and their positioning close to the MTOC, probably due to their role in phagosomal membrane stability and the connection of the SCV with motor proteins.
Discussion
This work identifies a novel phenomenon involving autophagy that is intimately linked to a persistent infection by an intracellular bacterial pathogen. Our data support a mechanism of pathogen control based on digestion of only some members of the intracellular bacterial progeny. Following entry into the fibroblast, S. Typhimurium induces a compacted aggregate formed by host endomembranes of endo/lysosomal origin. This aggregate is, in turn, responsible of the reduction in the intracellular bacterial progeny. Thus, aggrephagy eliminates both the aggregate and some nearby SCV that are in the same autophagosome and, as a consequence, are “incidentally” engulfed and digested within this compartment. To our knowledge, this is the first example of a phenomenon involving “simultaneous” selective macroautophagy of 2 different entities: whole bacteria and a membranous aggregate composed of host material. Fortuitous elimination of intracellular bacteria by this mechanism, which takes place at a rather late infection time (12 to 24 h), appears to affect pathogen fate selectively. Thus, not all the SCV are entrapped within VIM cages and such selection is reflected by their escape from the aggresome area. As it was captured by our live cell imaging experiments (Fig. S11), the phenomenon illustrates a “race against the clock” with the pathogen attempting to avoid ingestion by the autophagosome. In this scenario, it might occur that only those SCV physically connected to the membranous aggresome at the time that the phagophore membrane is completely sealed, are those SCV that become captured and digested by the fibroblast. The requirement of MT dynamics for formation of the LGP+ aggregate provides in our opinion a strong evidence for differentiating this aggresome from the ALIS structures that are reported by Mesquita et al. in epithelial cells infected with S. Typhimurium.Citation59 We also confirmed that incubation of fibroblasts with an extract of heat-killed bacteria (which contains lipopolysaccharide [LPS] and flagellin) does not trigger formation of the LGP+ aggresome (Fig. S12). The phenotype displayed by the ssaVΔ mutant, which does not induce LGP+ aggregates but releases LPS inside the infected fibroblasts (Fig. S12), supports also the notion of an aggrephagy event that occurs independently of TLR signaling. This is in marked contrast to the formation of ALIS by S. Typhimurium in epithelial cells.Citation59
Live cell-imaging microscopy allowed us to uncover how the distinct compartments that interact with the autophagy machinery evolve in the S. Typhimurium-infected fibroblast. Besides the distinct structural features observed, we identified differences with epithelial cells regarding autophagy receptors, timing of the autophagy events and the pathogen functions involved. The epithelial cell harboring S. Typhimurium stimulates xenophagy via the recognition of danger signals as ubiquitinated host or bacterial componentsCitation24 and LGALS8 bound to glycans exposed in damaged SCV.Citation28 Autophagy machinery is recruited to these sites by CALCOCO2, SQSTM1, OPTN, and CALCOCO3, the latter a CALCOCO2 paralog.Citation23,30,31 In fibroblasts, we were unable to detect ubiquitin in the LGP+ aggregate. Moreover, while in epithelial cells S. Typhimurium uses deubiquitinases like SseL to limit selective autophagy against ALIS,Citation59 our study in fibroblasts shows that the pathogen translocates SseL but that this SPI2 effector is not responsible for the lack of ubiquitin in the LGP+ aggregate. SseL must therefore play another yet-unknown role in the persistent infection of fibroblasts. Similarly, we did not detect CALCOCO2 in the LGP+-aggregate. The absence of this autophagy receptor might indicate that the tubular-vesicular elements that compact to form the membranous aggresome retain membrane integrity. Whether the molecular determinants for phagophore formation around the LGP+ aggregate and the nearby SCV respond to signals in the membrane of the “collapsed-SIF” or, alternatively, to the dynamic properties of the aggresome itself, remain to be investigated. According to our FRAP data, we favor the idea of recognition by the autophagy machinery based on the insoluble state of the LGP+ aggregate, as it was proposed for proteinaceous inclusions.Citation54 Future studies on other autophagy components very recently associated with S. Typhimurium clearance, such as MYO6 (myosin VI) and the autophagy receptor CALCOCO3,Citation31 might also shed light into the mechanism responsible for the aggrephagy event described here.
Presence of diacylglycerol (DAG) in the SCV, thought to be associated with membrane damage, also induces S. Typhimurium xenophagy.Citation32 Localization of DAG to SCV depends on the SPI1-T3SSCitation32 whereas our data in fibroblasts showed that this secretion system is dispensable for the aggrephagy event. A recent report provides evidence for a second role of autophagy in mediating sealing of the SCV membrane damaged by activity of the SPI1-T3SS, a phenomenon required for subsequent induction of the SPI2-T3SS.Citation33 Since we observed aggrephagy at the same extent in cells infected with wild-type and SPI1Δ strains () and SPI2-T3SS is required for the formation of the LGP+ aggresome (), we conclude that in fibroblasts SPI2-T3SS might be normally activated by most wild-type bacteria in the absence of a functional SPI1-T3SS.
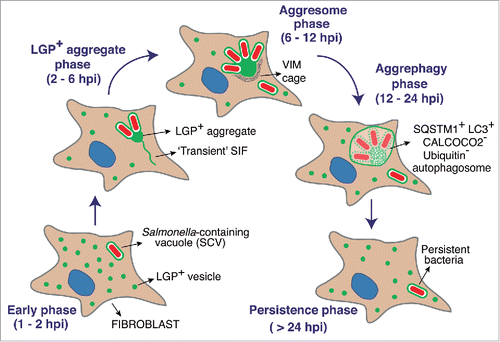
Our study also uncovered differences in the temporal execution of the distinct autophagy processes identified. Whereas S. Typhimurium xenophagy occurs early in the infection (1 to 2 hpi) in both epithelial cells and fibroblasts, aggrephagy of LGP+ membranous aggregates and nearby SCV takes place at medium-late infection times (from 6 to 8 hpi). Thus, xenophagy and aggrephagy are not alternative but coexisting mechanisms in the fibroblast, which supports the idea of fully independent autophagy machineries. These temporal differences could reflect distinct pathogen activities as the infection progresses. SPI1-T3SS might favor early recognition of S. Typhimurium by autophagy whereas our data point to a later contribution of SPI2-T3SS to the formation of the LGP+ membranous aggregate. SIF-like transient filaments are observed in the fibroblast at 2 to 6 hpi, a time at which SPI2 effectors such as SifA, SseJ and SopD2 could facilitate SCV membrane dynamics and generation of filamentous membrane extensions. The reasons for the subsequent collapse in this membrane dynamics are unknown. A plausible explanation could be a change in the prolife or levels of SPI2 effectors used by intracellular bacteria. Our previous data show that intracellular S. Typhimurium produces and translocates SPI2 effectors such as SseJ, PipB, and PipB2 at late infection times, 24 hpi.Citation60 At this late time, the LGP+ aggregate has formed and has been digested by aggrephagy (). Thus, the data obtained at 8 to 16 hpi with the single and double mutants lacking SifA, SseJ or SopD2 () are consistent with 2 temporally differentiated actions of SPI2 effectors. A first wave dedicated to trigger aggrephagy (up to 16 hpi) and, the second, to stabilize an intracellular lifestyle compatible with a long-lasting persistence state (> 24 hpi). This latter stage is consistent with the requirement of SPI2-T3SS for the maintenance of S. Typhimurium viability at late infection times (≥ 24 hpi) shown in fibroblasts.Citation16 depicts a model that integrates the data obtained in the fibroblast infection model and emphasizes the autophagy phases defined by live-cell imaging microscopy. This model highlights the link between aggrephagy of the membranous LGP+ aggregate and the establishment of a persistent state by a bacterial population that avoids destruction within the autophagosome. This subversion of selective autophagy by S. Typhimurium in fibroblasts represents an efficient strategy to establish a persistent infection. Our study may also favor future investigation to increase our still limited knowledge of the molecular mechanisms underlying macroautophagy of aggregates.
Figure 10. Model depicting how aggrephagy of host endomembranes might be exploited by S. Typhimurium to control bacterial progeny and establish an intracellular persistent infection. The model shows successive phases based on single cell and live-cell imaging microscopy analyses described in this study and performed over a 24-h infection period. Note the late time (after ∼12 hpi) at which aggrephagy takes place.
Materials and methods
Bacterial strains and plasmids
The S. enterica serovar Typhimurium strain SV5015 used in this study, described in the fibroblast infection model,Citation18 is a His+ derivative of the mouse virulent strain SL1344.Citation61 Derivative SV5015 strains were generated bearing plasmids pC.IGdsRed33 and pFPVmcherryCitation38 for constitutive production of fluorescent proteins DsRed and mCherry, respectively. The S. Typhimurium isogenic strains used include: SV5015 (wild type); MD1156 (pipB::3xFlag-Km); MD1810 (SV5015/pC.IGdsRed); MD2441 (SV5015/pFPVmcherry); MD3604(SPI1Δ);Citation62 MD2286 (SPI1Δ/pFPVmcherry); MD3601 (ssaVΔ), MD2500 (ssaVΔ/pFPVmcherry); MD3995 (sseLΔ); MD3996 (sseL::3xFlag-Km); MD2301 (steAΔ); MD4203 (steA::3xFlag-Km);Citation63 MD3622 (sseJΔ/pC.IGdsRed); MD3623 (sifAΔ/pC.IGdsRed); MD3624 (sopD2Δ/pC.IGdsRed); MD4707 (sifAΔ sseJΔ/pC.IGdsRed); and, MD4798 (sifAΔ sopD2Δ/pC.IGdsRed). Bacteria were grown in Luria broth, LB (Affymetrix, 75852) at 37°C. When appropriate, medium was supplemented with kanamycin (30 μg/ml), ampicillin (50 μg/ml) or chloramphenicol (10 μg/ml).
Bacterial infection of eukaryotic cells
NRK-49F rat fibroblasts (American Type Culture Collection, CRL-1570) and BJ-5ta human fibroblasts (American Type Culture Collection, CRL-4001) were used as cell models in which S. Typhimurium persists with limited proliferation capacity.Citation16,60 The S. Typhimurium-fibroblast infection model is also characterized by the lack of cytotoxicity in the infected cell for long time periods.Citation41 HeLa human epithelial cells (American Type Culture Collection, CCL-2) were used as control host cell type in which S. Typhimurium proliferates actively. These eukaryotic cell lines were infected with S. Typhimurium wild-type strain SV5015 or isogenic mutant strains, as described.Citation62 For live cell imaging microscopy, stable transfectants expressing CD63-GFP were generated from NRK-49F and HeLa cells using geneticin (ThermoFisher Scientific, 11811023) to select cells bearing mammalian CD63-GFP plasmid and following the Lonza technical guide (http://www.lonza.com/). The software packages used for image processing were ImageJ 64-bit 1.48v (Wayne Rasband, US National Institutes of Health), LAS AF 2.6.0 and 3.1.0 (Leica Microsystems), Metamorph offline 64-bit 7.8.8.0 (Molecular Devices), Imaris 7.7.1 (Bitplane Oxford Instruments) and Adobe Illustrator CS5 15.1.0 (Adobe Systems Inc.).
Antibodies and fluorescent dyes
The mouse monoclonal anti-human LAMP1 (clone H4A3), -human LAMP2 (clone H4B4) and -human CD63 (clone H5C6) antibodies (mAb) were from the Developmental Studies Hybridoma Bank (DSHB, University of Iowa, Iowa City, IA, USA). Other mouse mAb used recognize S. Typhimurium lipopolysaccharide (LPS; clone MLK33; a gift of J.M. Slauch, University of Illinois, Champaign-Urbana, IL, USA), rat LAMP2 lysosomal integral protein (clone 5G10; a gift of J. S. Bonifacino, NIH, Bethesda, MD, USA), rat SCARB2 (a gift of I. Sandoval, CBM Severo Ochoa, Madrid, Spain), mono- and poly-ubiquitinated conjugates (Enzo Life Sciences, BML-PW8810-0100), phagophore and autophagosome protein LC3B isoform (NanoTools, 0231-100/LC3-5F10), vacuolar ATPase (a gift of S.B. Sato, Kyoto University, Japan), Flag epitope (Sigma-Aldrich, F3165-5MG), pig VIM (vimentin; Sigma-Aldrich, MAB3400), and E. coli DnaK (Enzo Life Sciences, ADI-SPA-880-D). We also used rabbit polyclonal anti-S. Typhimurium LPS (Difco Laboratories, 2948-47-6), -human CALCOCO2 (a gift of J. Kendrick-Jones, MRC Laboratory of Molecular Biology, UK), -human CTSD (cathepsin D; Dako, A0561) and -human SQSTM1 (Abcam, ab56416) antibodies. As secondary fluorescent probes, we used goat anti-mouse and goat anti-rabbit antibodies conjugated to Alexa Fluor 488 nm, 546 nm, 594 nm and 647 nm (Life Technologies, A-11029, A-11034, A-11030, A-11035, A-11032, A-11037, A-21235 and A21245), the lysosome probe LysoTracker Deep Red (Life Technologies, A-22285), Alexa Fluor 660 conjugated to the F-actin probe phalloidin (Life Technologies, W-32466), and Alexa Fluor 647 conjugated to the trans-Golgi-derived vesicle marker wheat germ agglutinin (WGA; Life Technologies, L-7528). As secondary antibodies for western blot, we used goat anti-mouse or -rabbit antibody conjugated to horseradish peroxidase at 1:5000 (Bio-Rad, 1706516 and 176515). Blots were developed with ECL reagent (GE Healthcare Life Sciences, RPN2232). As a DNA marker, we used DAPI (4′-6′- diamidino-2-phenylindole) (Sigma-Aldrich, D9542).
Incubation of eukaryotic cells with extracts of killed-inactivated bacteria
dsRed-expressing S. Typhimuirum wild-type strain was grown overnight in LB (Affymetrix, 75852) at 37°C with agitation. One ml of this culture was subjected to 3 boiling (5 min at 95°C) and freezing (5 min at −80°C) cycles. NRK-49F cells expressing CD63-GFP were incubated for 4 h with 250 μl of the extract containing heat- and freeze-inactivated bacteria plus 250 μl of DMEM (ThermoFisher Scientific, 52100-021). NRK49F fibroblasts exposed to these extracts were fixed and processed for fluorescence microscopy to visualize CD63 distribution. Absence of red signal served as a control for the killing efficiency.
Starvation assay for autophagy induction
Infection of NRK-49F rat fibroblasts and determination of viable intracellular bacteria were as described.Citation62 To induce starvation, infected fibroblasts were incubated from 4 to 8 hpi in Hanks buffered salt solution (ThermoFisher Scientific, 24020133) containing 1% regular tissue culture medium, as described.Citation42 The cells were further maintained in regular medium (DMEM with 10% fetal bovine serum [FBS]).
Western blot analyses
3 × 107 NRK-49F fibroblasts were infected with isogenic S. Typhimurium steA::3xFlag, sseL::3xFlag and pipB::3xFlag tagged strains. At 8 hpi, fibroblasts were detached by scraping, collected by centrifugation (250 × g, 10 min, 4°C) and resuspended in 100 μl of Laemmli sample buffer (87.5 mM Tris-HCl, pH 6.8, 3.2% [w/v] SDS, 7.5% [v/v] glycerol, 150 mM DTT). For subcellular fractionation, cells collected after centrifugation were resuspended in 100 μl of TEN buffer (0.1 M Tris-HCl, pH 8, 0.01 M EDTA, pH 8, 1 M NaCl) with 1 % (v/v) Tween 20 (Sigma-Aldrich, P1379) and protease inhibitors (Roche, 11873580001). These cell suspensions were centrifuged (4,000 × g, 10 min, 4°C). Pellet (nuclei and bacteria) and supernatant fractions (cytosolic fractions) were resuspended and adjusted to a final volume of 100 μl of Laemmli sample buffer. SDS-PAGE and western blot were as described,Citation63 using anti-Flag as primary antibody. Bacterial DnaK was used to control protein loading and the subcellular fractionation procedure. Fibroblasts infected with untagged S. Typhimurium wild type strain and uninfected fibroblasts served as additional controls.
Immunofluorescence microscopy in fixed cells
Fibroblasts or epithelial cells infected with S. Typhimurium were fixed postinfection at the indicated times in 4% paraformaldehyde (Sigma-Aldrich, 158127) (20 min, room temperature), then processed for immunofluorescence microscopy as described.Citation60 Coverslips were mounted on glass slides using ProLong (Life Technologies, P36930). For epifluorescence microscopy, we used an inverted Leica DMI 6000B microscope with an automated CTR/7000 HS controller (Leica Microsystems, Wetzlar, Germany) and an Orca-R2 CCD camera (Hamamatsu Photonics, Hamamatsu, Japan), and for optical sectioning, a Leica TCS SP5 spectral laser scanning confocal microscope (Leica Microsystems, Wetzlar, Germany).
Live-cell imaging microscopy
Stably transfected NRK-49F or HeLa cells expressing CD63-GFP were used for video-microscopy experiments. Cells were cultured in 4-well dishes (Ibidi, 80426) and maintained in Fluorobrite DMEM (ThermoFisher Scientific, A1896701) supplemented with 10% FBS (37°C, 5% CO2) for the duration of time-lapse image acquisition. Cells were infected with the indicated S. Typhimurium strain that expressed mCherry or DsRed protein, as described.Citation38 In experiments recorded for 2 to 24 hpi, the rate was 30 min/frame for the first 6 h and 1 h/frame for the remainder 18 h, with multi-position mode and 2 × 2 binning. In experiments lasting up to 10 min, the rate was 30 s/frame with no binning. Images were acquired in an inverted Leica DMI 6000B microscope with an in vivo stage and a CTR/7000 HS controller (Leica Microsystems, Germany), and an Orca-R2 CCD camera (Hamamatsu Photonics, Japan).
Correlative light and electron microscopy (CLEM)
CLEM was used to identify precisely the location of S. Typhimurium-induced LGP+ aggregates. For definitive selection of regions of interest for ultrastructural studies, we identified spherical membranous aggregates surrounded by SCV. Stable NRK-49F transfectants expressing CD63-GFP were cultured on gridded coverslips (Electron Microscopy Sciences, 72265-50) and infected with a mCherry-expressing S. Typhimurium strain. At 8 hpi, coverslips were mounted in a cell chamber for microscopy (ThermoFisher Scientific, A7816) using Fluorobrite DMEM-10% FBS (37°C, 5% CO2) for the duration of image acquisition. LGP+-aggregates were positioned in coordinates based on the numbered pattern on the coverslip. Cells of interest harboring LGP+-aggregates were fixed in 1% glutaraldehyde (TAAB Laboratories, G010) (1 h, room temperature) and with 1% osmium tetroxide (TAAB Laboratories, O015), 0.8% potassium ferricyanide (TAAB Laboratories, P018) (1 h, 4°C). After washing, cells were dehydrated in a concentrated acetone (Meck Millipore, 1002990500) series (4°C). After overnight incubation in TAAB-812 epoxy resin (TAAB Laboratories Equipment, T024) in acetone (1:1), cells were infiltrated in successive steps in pure resin for 2 d. Resin was polymerized (60°C, 2 d) and ultrathin serial sections (50 to 70 nm) parallel to the coverslip base were obtained in a Leica EM UC6 ultramicrotome (Leica Microsystems, Wetzlar, Germany), placed over slot copper grids (Electron Microscopy Sciences, G2010-Cu) covered with a layer of 0.5% formvar (TAAB Laboratories, F145/100), dried, and stained with saturated uranyl acetate (Electron Microscopy Sciences, 541-09-03) and lead citrate (Fluka, 2081411). Samples were studied in a JEM 1011 electron microscope (JEOL, Peabody, MA, USA) at 100 kV.
Fluorescence recovery after photobleaching (FRAP) assays
For FRAP analyses of individual SCV, stable NRK-49F transfectant fibroblasts expressing CD63-GFP were infected with S. Typhimurium MD2441 (WT/pFPVmcherry), MD2286 (SPI1Δ/pFPVmcherry) or, MD2500 (ssaVΔ/pFPVmcherry) strains. FRAP assays began at 3 hpi. Individual SCV were bleached at 488-nm laser wavelength and tracked by differential interference contrast (DIC) microscopy. Fluorescence recovery was monitored by capture of images every 30 s for 7.5 min. For FRAP analysis of LGP+-aggregates, stable NRK-49F transfectant fibroblasts expressing CD63-GFP were infected with DsRed-expressing S. Typhimurium MD1810 (WT/pC.IGdsRed). FRAP assays began at 8 hpi, when the central region of the LGP+-aggregate was bleached. Fluorescence recovery was monitored by capturing images every 10 s for 2.5 min. Images were acquired with a Leica TCS SP5 spectral laser scanning confocal microscope (Leica Microsystems, Germany). Mean fluorescence intensity of selected regions of interest was measured with ImageJ (US National Institutes of Health) and the relative fluorescence intensity (RFI) was calculated as the mean of 10 data points. Plots were prepared with Prism 5.0 software (GraphPad Inc.).
Quantification of aggregate size and subcellular location of LGP+ aggregates
Images were acquired in the GFP, DsRed and DAPI channels in a wide-field fluorescence microscope (Leica DMI6000B) (Leica Microsystems, Wetzlar, Germany) using an Orca-R2 CCD camera (Hamamatsu Photonics, Japan). Parameters corresponding to “Distance aggregate-nucleus centroid” and “Aggregate surface” were calculated using ImageJ 64-bit 1.48v. Nuclei centroids were determined by applying an intensity threshold in the DAPI image obtaining particles corresponding to single nuclei. After analyzing the particles a number was shown in the centroid of each nucleus. Distance was measured between the center of the LGP+ aggregate to the nucleus centroid and expressed as microns. Aggregate surface was calculated by establishing a threshold in the GFP channel that included the entire aggregate surface, followed by analysis of the particles of interest. Surfaces are expressed in squared microns. At least one hundred LGP+ aggregates were examined per S. Typhimurium strain. Statistics for these analyses were performed using Prism version 5.0 (GraphPad Inc.). After discarding normal distribution of values with D'Agostino and Pearson normality test, nonparametric t test (Kruskal-Wallis test and applying Dunns post-hoc test) determined distribution of probabilities. Values were considered significant when the P value was below 0.05: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Selective digitonin permeabilization
NRK-49F fibroblasts and HeLa epithelial cells were infected with DsRed-expressing wild-type S. Typhimurium. Eukaryotic cells and bacteria were coincubated for 10 min followed by 3 washes with complete tissue culture medium. Infected cultures were kept at 37°C for 1, 4 or 8 h in the presence of 25 µg/ml gentamicin (ThermoFisher Scientific, 15750060). At these infection times, cells were treated with 50 µg/ml digitonin (Sigma, D141) for 30 sec and subsequently fixed with 4% paraformaldehyde (Sigma-Aldrich, 158127) for 20 min. These cells were stained with the following primary antibodies: rabbit polyclonal anti-S. Typhimurium LPS (Difco Laboratories, 2948-47-6); and, mouse monoclonal antibodies 5G10 (gift of J.S. Bonifacino) and H4A3 (Developmental Studies Hybridoma Bank, H4A3), which recognize rat LAMP2 lysosomal membrane glycoprotein and the human lysosomal glycoprotein LAMP1, respectively. Secondary antibodies included anti-mouse and anti-rabbit goat antibodies conjugated to Alexa Fluor 488 and Alexa Fluor 647, respectively (ThermoFisher Scientific, A-11029 and A-21245). Nuclei were stained with DAPI. Images were acquired in a Leica TCS SP5 confocal microscope (Leica Microsystems, Germany). Cytosolic bacteria were considered those stained with the anti-LPS antibody but lacking LGP-specific labeling (5G10, H4A3).
Statistical analysis
Data were analyzed by a 2-tailed Student t test using Prism 5.0 software. Differences with P <0.001 were considered highly significant and P ≥ 0.05, not significant (ns).
Abbreviations
| ALIS | = | aggresome-like induced structures |
| CALCOCO2/NDP52 | = | calcium binding and coiled-coil domain 2 |
| CALCOCO3/TAX1BP1 | = | calcium binding and coiled-coil domain 3 |
| CD63/LAMP-3 | = | CD63 molecule (238 amino acids, distinct from LAMP3/CD208 of 416 amino acids) |
| CLEM | = | correlative light and electron microscopy |
| DAG | = | diacylglycerol |
| DsRed | = | Discosoma sp. red fluorescent protein |
| DALIS | = | dendritic cell aggresome-like induced structures |
| FRAP | = | fluorescence recovery after photo-bleaching |
| GFP | = | green fluorescent protein |
| LAMP1/CD107a | = | lysosomal-associated membrane protein 1 |
| LAMP2/CD107b | = | lysosomal-associated membrane protein 2 |
| LGALS8/galectin-8 | = | lectin, galactoside binding, soluble, 8 |
| LGP | = | lysosomal membrane glycoprotein |
| MAP1LC3/LC3 | = | microtubule-associated protein 1 light chain 3 |
| MTOC | = | microtubule organizing center |
| OPTN | = | optineurin |
| PE | = | phosphatidylethanolamine |
| SCARB2/Limp-II | = | scavenger receptor class B member 2 |
| SCV | = | Salmonella-containing vacuole |
| SIF | = | Salmonella-induced filament |
| SNCAIP/synphilin-1 | = | synuclein α interacting protein |
| SPI1 | = | Salmonella-pathogenicity island 1 |
| SPI2 | = | Salmonella pathogenicity island 2 |
| SQSTM1/p62 | = | sequestosome 1 |
| T3SS | = | type-III protein secretion system |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KAUP_A_1208888_Supplementary_material.zip
Download Zip (56.1 MB)Acknowledgments
We thank Gillian Griffiths (Cambridge Institute for Medical Research, University of Cambridge, UK) for the CD63-GFP construct; John Kendrick-Jones (MRC Laboratory of Molecular Biology, Cambridge, UK) for anti-CALCOCO2 antibody; Satoshi B. Sato (Kyoto University, Japan) for anti-vATPase antibody; Juan S. Bonifacino (NIH, Bethesda, MD, USA) for the 5G10 monoclonal antibody; Ignacio Sandoval (CBM Severo Ochoa, Madrid, Spain) for anti-SCARB2 antibody; James M. Slauch, (University of Illinois, IL, USA) for anti-S. Typhimurim LPS antibody; Dirk Bumann (Biozentrum, University of Basel, Switzerland) for the pC.IGdsRed plasmid; Olivia Steele-Mortimer (Rocky Mountain Labs, NIAID/NIH, MT, USA) for the pFPVmcherry plasmid; Francisco Ramos-Morales (University of Seville, Spain) for the S. Typhimurium steAΔ and steA::3xFlag strains; Sylvia Gutierrez-Erlandsson for technical assistance at the CNB Confocal Microscopy Unit; and, Catherine Mark for editorial assistance. We are also grateful to Jost Enninga (Pasteur Institute, Paris, France) for facilitating lab space, reagents and access to microscopes.
Funding
This work was supported by grants BIO2013-46281P, CSD2008/00013, and IPT2012-0213-060000 (to FGdP) from the Spanish Ministry of Economy and Competitiveness.
Related Research Data
References
- Rogov V, Dotsch V, Johansen T, Kirkin V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell 2014; 53:167-78; PMID:24462201; http://dx.doi.org/10.1016/j.molcel.2013.12.014
- Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol 2013; 13:722-37; PMID:24064518; http://dx.doi.org/10.1038/nri3532
- Jo EK, Yuk JM, Shin DM, Sasakawa C. Roles of autophagy in elimination of intracellular bacterial pathogens. Front Immunol 2013; 4:97; PMID:23653625; http://dx.doi.org/10.3389/fimmu.2013.00097
- Bradfute SB, Castillo EF, Arko-Mensah J, Chauhan S, Jiang S, Mandell M, Deretic V. Autophagy as an immune effector against tuberculosis. Curr Opin Microbiol 2013; 16:355-65; PMID:23790398; http://dx.doi.org/10.1016/j.mib.2013.05.003
- Choy A, Dancourt J, Mugo B, O'Connor TJ, Isberg RR, Melia TJ, Roy CR. The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science 2012; 338:1072-6; PMID:23112293; http://dx.doi.org/10.1126/science.1227026
- Ogawa M, Yoshikawa Y, Mimuro H, Hain T, Chakraborty T, Sasakawa C. Autophagy targeting of Listeria monocytogenes and the bacterial countermeasure. Autophagy 2011; 7:310-4; PMID:21193840; http://dx.doi.org/10.4161/auto.7.3.14581
- Dortet L, Mostowy S, Samba-Louaka A, Gouin E, Nahori MA, Wiemer EA, Dussurget O, Cossart P. Recruitment of the major vault protein by InlK: a Listeria monocytogenes strategy to avoid autophagy. PLoS Pathog 2011; 7:e1002168; PMID:21829365; http://dx.doi.org/10.1371/journal.ppat.1002168
- Ogawa M, Yoshikawa Y, Kobayashi T, Mimuro H, Fukumatsu M, Kiga K, Piao Z, Ashida H, Yoshida M, Kakuta S, et al. A Tecpr1-dependent selective autophagy pathway targets bacterial pathogens. Cell Host Microbe 2011; 9:376-89; PMID:21575909; http://dx.doi.org/10.1016/j.chom.2011.04.010
- Rytkonen A, Poh J, Garmendia J, Boyle C, Thompson A, Liu M, Freemont P, Hinton JC, Holden DW. SseL, a Salmonella deubiquitinase required for macrophage killing and virulence. Proc Natl Acad Sci U S A 2007; 104:3502-7; PMID:17360673; http://dx.doi.org/10.1073/pnas.0610095104
- Deretic V, Kimura T, Timmins G, Moseley P, Chauhan S, Mandell M. Immunologic manifestations of autophagy. J Clin Invest 2015; 125:75-84; PMID:25654553; http://dx.doi.org/10.1172/JCI73945
- Malik-Kale P, Jolly CE, Lathrop S, Winfree S, Luterbach C, Steele-Mortimer O. Salmonella - at home in the host cell. Front Microbiol 2011; 2:125; PMID:21687432; http://dx.doi.org/10.3389/fmicb.2011.00125
- Bakowski MA, Braun V, Brumell JH. Salmonella-containing vacuoles: directing traffic and nesting to grow. Traffic 2008; 9:2022-31; PMID:18778407; http://dx.doi.org/10.1111/j.1600-0854.2008.00827.x
- Garcia-del Portillo F, Nunez-Hernandez C, Eisman B, Ramos-Vivas J. Growth control in the Salmonella-containing vacuole. Curr Opin Microbiol 2008; 11:46-52; PMID:18282735; http://dx.doi.org/10.1016/j.mib.2008.01.001
- Watson KG, Holden DW. Dynamics of growth and dissemination of Salmonella in vivo. Cell Microbiol 2010; 12:1389-97; PMID:20731667; http://dx.doi.org/10.1111/j.1462-5822.2010.01511.x
- Gog JR, Murcia A, Osterman N, Restif O, McKinley TJ, Sheppard M, Achouri S, Wei B, Mastroeni P, Wood JL, et al. Dynamics of Salmonella infection of macrophages at the single cell level. J R Soc Interface 2012; 9:2696-707; PMID:22552918
- Cano DA, Martinez-Moya M, Pucciarelli MG, Groisman EA, Casadesus J, Garcia-Del Portillo F. Salmonella enterica serovar Typhimurium response involved in attenuation of pathogen intracellular proliferation. Infect Immun 2001; 69:6463-74; PMID:11553591; http://dx.doi.org/10.1128/IAI.69.10.6463-6474.2001
- Martinez-Moya M, de Pedro MA, Schwarz H, Garcia-del Portillo F. Inhibition of Salmonella intracellular proliferation by non-phagocytic eucaryotic cells. Res Microbiol 1998; 149:309-18; PMID:9766231; http://dx.doi.org/10.1016/S0923-2508(98)80436-1
- Nunez-Hernandez C, Tierrez A, Ortega AD, Pucciarelli MG, Godoy M, Eisman B, Casadesús J, García-del Portillo F. Genome expression analysis of nonproliferating intracellular salmonella enterica serovar typhimurium unravels an acid pH-dependent PhoP-PhoQ response essential for dormancy. Infect Immun 2013; 81:154-65; PMID:23090959; http://dx.doi.org/10.1128/IAI.01080-12
- Yu HB, Croxen MA, Marchiando AM, Ferreira RB, Cadwell K, Foster LJ, Finlay BB. Autophagy facilitates Salmonella replication in HeLa cells. MBio 2014; 5:e00865-14; PMID:24618251
- Holzer SU, Hensel M. Divergent roles of Salmonella pathogenicity island 2 and metabolic traits during interaction of S. enterica serovar typhimurium with host cells. PLoS One 2012; 7:e33220; PMID:22427996; http://dx.doi.org/10.1371/journal.pone.0033220
- Schroeder N, Mota LJ, Meresse S. Salmonella-induced tubular networks. Trends Microbiol 2011; 19:268-77; PMID:21353564; http://dx.doi.org/10.1016/j.tim.2011.01.006
- Liss V, Hensel M. Take the tube: remodelling of the endosomal system by intracellular Salmonella enterica. Cell Microbiol 2015; 17:639-47; PMID:25802001; http://dx.doi.org/10.1111/cmi.12441
- Gomes LC, Dikic I. Autophagy in antimicrobial immunity. Mol Cell 2014; 54:224-33; PMID:24766886; http://dx.doi.org/10.1016/j.molcel.2014.03.009
- Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol 2009; 10:1215-21; PMID:19820708; http://dx.doi.org/10.1038/ni.1800
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131-45; PMID:17580304; http://dx.doi.org/10.1074/jbc.M702824200
- Zheng YT, Shahnazari S, Brech A, Lamark T, Johansen T, Brumell JH. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol 2009; 183:5909-16; PMID:19812211; http://dx.doi.org/10.4049/jimmunol.0900441
- Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011; 333:228-33; PMID:21617041; http://dx.doi.org/10.1126/science.1205405
- Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, Randow F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 2012; 482:414-8; PMID:22246324; http://dx.doi.org/10.1038/nature10744
- Verlhac P, Gregoire IP, Azocar O, Petkova DS, Baguet J, Viret C, Faure M. Autophagy receptor NDP52 regulates pathogen-containing autophagosome maturation. Cell Host Microbe 2015; 17:515-25; PMID:25771791; http://dx.doi.org/10.1016/j.chom.2015.02.008
- Narayanan LA, Edelmann MJ. Ubiquitination as an efficient molecular strategy employed in salmonella infection. Front Immunol 2014; 5:558; PMID:25505465; http://dx.doi.org/10.3389/fimmu.2014.00558
- Tumbarello DA, Manna PT, Allen M, Bycroft M, Arden SD, Kendrick-Jones J, Buss F. The autophagy receptor TAX1BP1 and the molecular motor myosin VI Are required for clearance of salmonella typhimurium by autophagy. PLoS Pathog 2015; 11:e1005174; PMID:26451915; http://dx.doi.org/10.1371/journal.ppat.1005174
- Shahnazari S, Namolovan A, Klionsky DJ, Brumell JH. A role for diacylglycerol in antibacterial autophagy. Autophagy 2011; 7:331-3; PMID:21079417; http://dx.doi.org/10.4161/auto.7.3.14045
- Kreibich S, Emmenlauer M, Fredlund J, Ramo P, Munz C, Dehio C, Enninga J, Hardt WD. Autophagy Proteins Promote Repair of Endosomal Membranes Damaged by the Salmonella Type Three Secretion System 1. Cell Host Microbe 2015; 18:527-37; PMID:26567507; http://dx.doi.org/10.1016/j.chom.2015.10.015
- Garcia-del Portillo F, Zwick MB, Leung KY, Finlay BB. Salmonella induces the formation of filamentous structures containing lysosomal membrane glycoproteins in epithelial cells. Proc Natl Acad Sci U S A 1993; 90:10544-8; PMID:8248143; http://dx.doi.org/10.1073/pnas.90.22.10544
- Birmingham CL, Smith AC, Bakowski MA, Yoshimori T, Brumell JH. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J Biol Chem 2006; 281:11374-83; PMID:16495224; http://dx.doi.org/10.1074/jbc.M509157200
- Steele-Mortimer O. The Salmonella-containing vacuole: moving with the times. Curr Opin Microbiol 2008; 11:38-45; PMID:18304858; http://dx.doi.org/10.1016/j.mib.2008.01.002
- Knodler LA. Salmonella enterica: living a double life in epithelial cells. Curr Opin Microbiol 2015; 23:23-31; PMID:25461569; http://dx.doi.org/10.1016/j.mib.2014.10.010
- Drecktrah D, Levine-Wilkinson S, Dam T, Winfree S, Knodler LA, Schroer TA, Steele-Mortimer O. Dynamic behavior of Salmonella-induced membrane tubules in epithelial cells. Traffic 2008; 9:2117-29; PMID:18785994; http://dx.doi.org/10.1111/j.1600-0854.2008.00830.x
- Rajashekar R, Liebl D, Seitz A, Hensel M. Dynamic remodeling of the endosomal system during formation of Salmonella-induced filaments by intracellular Salmonella enterica. Traffic 2008; 9:2100-16; PMID:18817527; http://dx.doi.org/10.1111/j.1600-0854.2008.00821.x
- Krieger V, Liebl D, Zhang Y, Rajashekar R, Chlanda P, Giesker K, Chikkaballi D, Hensel M. Reorganization of the endosomal system in Salmonella-infected cells: the ultrastructure of Salmonella-induced tubular compartments. PLoS Pathog 2014; 10:e1004374; PMID:25254663; http://dx.doi.org/10.1371/journal.ppat.1004374
- Cano DA, Pucciarelli MG, Martinez-Moya M, Casadesus J, Garcia-del Portillo F. Selection of small-colony variants of Salmonella enterica serovar typhimurium in nonphagocytic eucaryotic cells. Infect Immun 2003; 71:3690-8; PMID:12819049; http://dx.doi.org/10.1128/IAI.71.7.3690-3698.2003
- Shang L, Chen S, Du F, Li S, Zhao L, Wang X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci U S A 2011; 108:4788-93; PMID:21383122; http://dx.doi.org/10.1073/pnas.1100844108
- Lamark T, Johansen T. Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int J Cell Biol 2012; 2012:736905; PMID:22518139; http://dx.doi.org/10.1155/2012/736905
- Svenning S, Johansen T. Selective autophagy. Essays Biochem 2013; 55:79-92; PMID:24070473; http://dx.doi.org/10.1042/bse0550079
- von Muhlinen N, Thurston T, Ryzhakov G, Bloor S, Randow F. NDP52, a novel autophagy receptor for ubiquitin-decorated cytosolic bacteria. Autophagy 2010; 6:288-9; PMID:20104023; http://dx.doi.org/10.4161/auto.6.2.11118
- Perrin AJ, Jiang X, Birmingham CL, So NS, Brumell JH. Recognition of bacteria in the cytosol of Mammalian cells by the ubiquitin system. Curr Biol 2004; 14:806-11; PMID:15120074; http://dx.doi.org/10.1016/j.cub.2004.04.033
- Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol 1998; 143:1883-98; PMID:9864362; http://dx.doi.org/10.1083/jcb.143.7.1883
- Garcia-Mata R, Gao YS, Sztul E. Hassles with taking out the garbage: aggravating aggresomes. Traffic 2002; 3:388-96; PMID:12010457; http://dx.doi.org/10.1034/j.1600-0854.2002.30602.x
- Johnston JA, Illing ME, Kopito RR. Cytoplasmic dynein/dynactin mediates the assembly of aggresomes. Cell Motil Cytoskeleton 2002; 53:26-38; PMID:12211113; http://dx.doi.org/10.1002/cm.10057
- Zaarur N, Meriin AB, Bejarano E, Xu X, Gabai VL, Cuervo AM, Sherman MY. Proteasome failure promotes positioning of lysosomes around the aggresome via local block of microtubule-dependent transport. Mol Cell Biol 2014; 34:1336-48; PMID:24469403; http://dx.doi.org/10.1128/MCB.00103-14
- Szeto J, Kaniuk NA, Canadien V, Nisman R, Mizushima N, Yoshimori T, Bazett-Jones DP, Brumell JH. ALIS are stress-induced protein storage compartments for substrates of the proteasome and autophagy. Autophagy 2006; 2:189-99; PMID:16874109; http://dx.doi.org/10.4161/auto.2731
- Canadien V, Tan T, Zilber R, Szeto J, Perrin AJ, Brumell JH. Cutting edge: microbial products elicit formation of dendritic cell aggresome-like induced structures in macrophages. J Immunol 2005; 174:2471-5; PMID:15728449; http://dx.doi.org/10.4049/jimmunol.174.5.2471
- Wileman T. Aggresomes and pericentriolar sites of virus assembly: cellular defense or viral design? Annu Rev Microbiol 2007; 61:149-67; PMID:17896875; http://dx.doi.org/10.1146/annurev.micro.57.030502.090836
- Wong E, Bejarano E, Rakshit M, Lee K, Hanson HH, Zaarur N, Phillips GR, Sherman MY, Cuervo AM. Molecular determinants of selective clearance of protein inclusions by autophagy. Nat Commun 2012; 3:1240; PMID:23212369; http://dx.doi.org/10.1038/ncomms2244
- Rajashekar R, Liebl D, Chikkaballi D, Liss V, Hensel M. Live cell imaging reveals novel functions of Salmonella enterica SPI2-T3SS effector proteins in remodeling of the host cell endosomal system. PLoS One 2014; 9:e115423; PMID:25522146; http://dx.doi.org/10.1371/journal.pone.0115423
- Domingues L, Holden DW, Mota LJ. The Salmonella effector SteA contributes to the control of membrane dynamics of Salmonella-containing vacuoles. Infect Immun 2014; 82:2923-34; PMID:24778114; http://dx.doi.org/10.1128/IAI.01385-13
- Beuzon CR, Meresse S, Unsworth KE, Ruiz-Albert J, Garvis S, Waterman SR, Ryder TA, Boucrot E, Holden DW. Salmonella maintains the integrity of its intracellular vacuole through the action of SifA. EMBO J 2000; 19:3235-49; PMID:10880437; http://dx.doi.org/10.1093/emboj/19.13.3235
- Ruiz-Albert J, Yu XJ, Beuzon CR, Blakey AN, Galyov EE, Holden DW. Complementary activities of SseJ and SifA regulate dynamics of the Salmonella typhimurium vacuolar membrane. Mol Microbiol 2002; 44:645-61; PMID:11994148; http://dx.doi.org/10.1046/j.1365-2958.2002.02912.x
- Mesquita FS, Thomas M, Sachse M, Santos AJ, Figueira R, Holden DW. The Salmonella deubiquitinase SseL inhibits selective autophagy of cytosolic aggregates. PLoS Pathog 2012; 8:e1002743; PMID:22719249; http://dx.doi.org/10.1371/journal.ppat.1002743
- Nunez-Hernandez C, Alonso A, Pucciarelli MG, Casadesus J, Garcia-del Portillo F. Dormant intracellular Salmonella enterica serovar Typhimurium discriminates among Salmonella pathogenicity island 2 effectors to persist inside fibroblasts. Infect Immun 2014; 82:221-32; PMID:24144726; http://dx.doi.org/10.1128/IAI.01304-13
- Hoiseth SK, Stocker BA. Aromatic-dependent Salmonella typhimurium are non-virulent and effective as live vaccines. Nature 1981; 291:238-9; PMID:7015147; http://dx.doi.org/10.1038/291238a0
- Aiastui A, Pucciarelli MG, Garcia-del Portillo F. Salmonella enterica serovar typhimurium invades fibroblasts by multiple routes differing from the entry into epithelial cells. Infect Immun 2010; 78:2700-13; PMID:20368348; http://dx.doi.org/10.1128/IAI.01389-09
- Cardenal-Munoz E, Ramos-Morales F. Analysis of the expression, secretion and translocation of the Salmonella enterica type III secretion system effector SteA. PLoS One 2011; 6:e26930; PMID:22046414; http://dx.doi.org/10.1371/journal.pone.0026930