
ABSTRACT
Mitochondrial autophagy or mitophagy is a key component of mitochondrial quality control, which is necessary to maintain cellular bioenergetics. Pancreatic islet β-cells, which release insulin in response to circulating blood glucose levels, are particularly susceptible to mitochondrial dysfunction due to their high metabolic activity and energy requirements for insulin processing, maturation, and secretion. Therefore, dysregulated mitophagy has drawn interest in the etiology of β-cell failure in diabetes. We demonstrate that the pivotal β-cell mitophagy regulator, CLEC16A, is an E3 ligase that forms a ubiquitin-dependent tripartite complex with RNF41/NRDP1 and USP8. Maintenance of the CLEC16A-RNF41-USP8 mitophagy complex is necessary for maximal cellular respiration and insulin secretion. Further, we observe that diabetogenic metabolic stressors, including elevated glucose and fatty acids, destabilize the CLEC16A-RNF41-USP8 complex and lead to β-cell apoptosis. Thus, the β-cell mitophagy pathway requires ubiquitin signals to stabilize the CLEC16A-RNF41-USP8 complex and maintain mitochondrial quality control.
Mitochondrial quality control is crucial to preserve cellular respiration and respond to mitochondrial damage. The mitochondrial quality control pathway includes a comprehensive quality assurance apparatus, comprised of proteases, protein repair systems, misfolded protein chaperones, oxidative stress responses, fission-fusion dynamics, and elimination of impaired proteins by mitochondrial-derived vesicles. Mitophagy, the selective clearance of damaged or dysfunctional mitochondria by autophagy, is also critical for mitochondrial quality control. Pancreatic β-cells are an ideal vehicle for the study of mitophagy, due to their dependence on mitochondrial respiration and sensitivity to mitochondrial damage. We previously determined that CLEC16A is a vital regulator of mitophagy in β-cells, yet its discrete molecular action in the mitophagy pathway is unknown. Therefore, we endeavored to clarify the specific function of CLEC16A in β-cell mitophagy.
Ubiquitination, a post-translational modification involving the addition of activated ubiquitin moieties to protein substrates by E3 ubiquitin ligases, is a necessary signal for activity during mitophagy. CLEC16A binds, stabilizes, and prevents the proteasomal degradation of the E3 ligase RNF41, which proceeds to ubiquitinate the mitophagy initiator PARK2/PARKIN (itself an E3 ligase). CLEC16A-RNF41 action on PARK2 leads to its proteasomal degradation, thus preventing unnecessary activation of mitophagy. Therefore, the complex interplay of ubiquitin signaling between these E3 ligases led us to hypothesize that ubiquitination of mitophagy regulators upstream of PARK2 is necessary for β-cell mitophagy. We initially investigated RNF41 ubiquitination via the selective E3 ligase inhibitor lenalidomide, which is a chemotherapeutic agent associated with new-onset diabetes in humans. We observed that lenalidomide reduces RNF41 ubiquitination in β-cells. Lenalidomide treatment impairs β-cell function in humans, as well as decreasing insulin secretion and mitochondrial respiration in mouse islets. Indeed, in vivo administration of lenalidomide in mice leads to glucose intolerance and increased localization of mitochondria within autophagosomes in β-cells. Further, flux studies reveal that the accumulation of mitochondria targeted to phagophores (autophagosome precursors) by lenalidomide is due to impaired flux through mitophagy. These initial studies suggested that RNF41 ubiquitination ensures mitochondrial clearance and is essential for β-cell function.
We next sought to elucidate the mechanism of CLEC16A regulation of RNF41 in context with other upstream mitophagy factors. We found that CLEC16A paradoxically enhances ubiquitination of RNF41, while promoting RNF41 stability. Interestingly, lenalidomide treatment decreases CLEC16A-mediated RNF41 ubiquitination. Moreover, lenalidomide does not additionally impair glucose-stimulated insulin secretion beyond intrinsic defects known to occur in CLEC16A-deficient islets, suggesting that its effects on β-cells are through the CLEC16A-pathway. The action of CLEC16A to stabilize RNF41 is in contrast to USP8, a deubiquitinase that removes degradative ubiquitin conjugates from RNF41. Utilizing gain- and loss-of-function approaches, we observed that USP8 and CLEC16A work independently, yet in a complementary fashion, to enhance RNF41 stability. This led us to question whether CLEC16A stabilizes and ubiquitinates RNF41 via endogenous E3 ligase activity or via recruitment of other factors. We utilized in vitro approaches to demonstrate that CLEC16A has intrinsic E3 ligase activity, and possesses the ability to self-ubiquitinate as well as directly ubiquitinate RNF41. Further, we observed that CLEC16A added K48-independent nondegradative ubiquitin linkages to RNF41. Thus, we identified CLEC16A as an E3 ligase that stabilizes RNF41 via nondegradative ubiquitination.
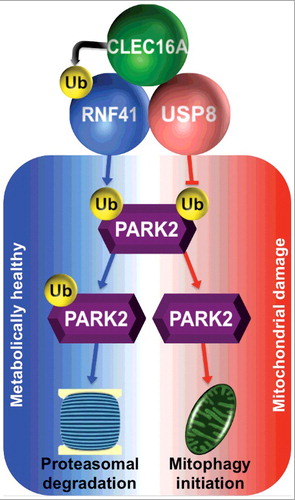
Beyond its role on RNF41 stability, USP8 has recently been shown to control mitophagy by removing K6 ubiquitin linkages from PARK2 to allow its mitochondrial localization. The convergence of 2 upstream mitophagy components (CLEC16A and USP8) on RNF41 stability and ubiquitination led us to hypothesize that these proteins coexist in a ubiquitin-dependent complex to modulate PARK2. Indeed, CLEC16A, RNF41, and USP8 colocalize and interact in a tripartite complex. Importantly, we observed that maintenance of the CLEC16A-RNF41-USP8 complex requires functional multimeric ubiquitination. We next interrogated the role of CLEC16A on USP8-mediated removal of K6 linkages from PARK2. Whereas CLEC16A acts in concert with USP8 to potentiate RNF41 stability, CLEC16A increases K6-linked ubiquitin on PARK2 and opposes the effects of USP8. We speculate that this tripartite complex restrains PARK2 activity during normal physiological states, which could then be tuned to ensure mitophagic flux following mitochondrial damage ().
Figure 1. Schema of CLEC16A-RNF41-USP8 complex action to integrate ubiquitin signals and fine-tune flux through mitophagy. Under physiological or metabolically healthy conditions (blue), CLEC16A assembles the upstream mitophagy complex and ubiquinates RNF41, leading to PARK2 targeting to the proteasome for degradation. Following increased mitochondrial damage (red), dissolution of the CLEC16A-RNF41-USP8 complex results in reduced levels of RNF41 and enhances USP8 action to deubiquitinate PARK2, thus allowing PARK2 mitochondrial localization to initiate mitophagy.
To investigate the importance of this complex under diabetogenic stimuli, we exposed human and rodent islets to high glucose concentrations and the fatty acid palmitate to simulate the hyperglycemia and hyperlipidemia (also known as glucolipotoxicity) observed in diabetes. We observed that the CLEC16A-RNF41-USP8 complex is destabilized by glucolipotoxic conditions in a CLEC16A-dependent manner, such that re-expression of CLEC16A can prevent dissolution of the complex and maintain RNF41 ubiquitination. A major downstream consequence of glucolipotoxicity in β-cells is apoptosis, and we found that overexpression of CLEC16A improves cell survival following glucolipotoxciity.
Taken together, we demonstrated that the E3 ligase CLEC16A modulates mitophagy through the coordinated action of a ubiquitin-dependent complex together with USP8 and RNF41. We surmise that this upstream complex fine-tunes mitophagic capacity through ubiquitination of both its own constituents and its effector PARK2. Given the importance of this complex in pancreatic β-cells, it may be a plausible candidate to protect β-cells from metabolic stress, apoptosis, and eventual diabetes progression. Understanding the actions of the CLEC16A-RNF41-USP8 complex in other metabolically active tissues and the physiological control of its ubiquitin linkage landscape will be valuable to potentially exploit the CLEC16A mitophagy pathway for translational benefits.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was reported by the authors
Acknowledgements
The authors acknowledge funding support from JDRF (CDA-2016-189 and SRA-2018-539), the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health (R01-DK-108921), the Brehm family, and the Anthony family. The JDRF Career Development Award to S.A.S. is partly supported by the Danish Diabetes Academy, which is supported by the Novo Nordisk Foundation.