
ABSTRACT
Circular RNAs (circRNAs) are highly expressed in the central nervous system and are involved in the regulation of physiological and pathophysiological processes. However, the potential role of circRNAs in stroke remains largely unknown. Here, using a circRNA microarray, we showed that circular RNA Hectd1 (circHectd1) levels were significantly increased in ischemic brain tissues in transient middle cerebral artery occlusion (tMCAO) mouse stroke models and further validated this finding in plasma samples from acute ischemic stroke (AIS) patients. Knockdown of circHectd1 expression significantly decreased infarct areas, attenuated neuronal deficits, and ameliorated astrocyte activation in tMCAO mice. Mechanistically, circHECTD1 functions as an endogenous MIR142 (microRNA 142) sponge to inhibit MIR142 activity, resulting in the inhibition of TIPARP (TCDD inducible poly[ADP-ribose] polymerase) expression with subsequent inhibition of astrocyte activation via macroautophagy/autophagy. Taken together, the results of our study indicate that circHECTD1 and its coupling mechanism are involved in cerebral ischemia, thus providing translational evidence that circHECTD1 can serve as a novel biomarker of and therapeutic target for stroke.
Abbreviations: 3-MA: 3-methyladenine; ACTB: actin beta; AIS: acute ischemic stroke; AS: primary mouse astrocytes; BECN1: beclin 1, autophagy related; BMI: body mass index; circHECTD1: circRNA HECTD1; circRNAs: circular RNAs; CBF: cerebral blood flow; Con: control; DAPI: 4ʹ,6-diamidino-2-phenylindole; ECA: external carotid artery; FISH: fluorescence in situ hybridization; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; Gdna: genomic DNA; GFAP: glial fibrillary acidic protein; GO: gene ontology; HDL: high-density lipoprotein; IOD: integrated optical density; LDL: low-density lipoprotein; LPA: lipoprotein(a); MAP1LC3B: microtubule-associated protein 1 light chain 3 beta; MIR142: microRNA 142; mNSS: modified neurological severity scores; MRI: magnetic resonance imaging; NIHSS: National Institute of Health Stoke Scale; OGD-R: oxygen glucose deprivation-reperfusion; PCR: polymerase chain reaction; PFA: paraformaldehyde; SQSTM1: sequestosome 1; TIPARP: TCDD inducible poly(ADP-ribose) polymerase; tMCAO: transient middle cerebral artery occlusion; TTC: 2,3,5-triphenyltetrazolium chloride; UTR: untranslated region; WT: wild type
Introduction
Circular RNAs (circRNAs), generated from back-spliced exons, have recently been identified as a naturally occurring family of non-coding RNAs that is highly represented in the eukaryotic transcriptome [Citation1]. These endogenous RNAs are characterized by a stable structure and high tissue-specific expression [Citation2]. CircRNAs are highly homologous and generally more stable than their linear counterparts because they lack accessible ends and are thus resistant to exonucleases [Citation2]. While most of the circRNAs reported so far have been exonic circRNAs, a class of intron-containing exonic circRNAs can be found predominantly in the nucleus [Citation3], where they promote transcription of their parental genes. Several classes of non-coding RNAs have been shown to be involved in the regulation of physiological and pathophysiological processes including development and the senescence, hypertrophy and failure of the heart as well as cell growth [Citation4–Citation7]. Despite the biological importance of circRNAs that has been revealed, little is known regarding their expression and biological function in the pathogenesis of stroke.
Stroke is a common neurological disorder and one of the major causes of permanent morbidity and disability worldwide [Citation8,Citation9]. Intravenous administration of recombinant tissue plasminogen activator is currently the only FDA approved therapy for acute ischemic stroke (AIS) patients. However, due to a narrow time window and the side effects of recombinant tissue plasminogen activator, the efficacy of ischemic stroke thrombolytic therapy is limited [Citation10–Citation12]. In the past few decades, the primary approach for stroke has focused on neuroprotection to rescue ischemic neurons in the brain from irreversible injury. Several clinical trials intended to identify effective therapies have been undertaken [Citation13]; however, despite showing efficacy in experimental stroke models, all of these efforts have failed to provide significant benefits in clinical stroke trials [Citation14]. Therefore, novel treatment approaches that enable clinicians and researchers to overcome the above side effects and to extend the therapeutic time windows of existing agents are urgently needed.
To develop an ideal therapy and broaden treatment targets, we must consider therapeutic approaches that benefit multiple cell types, which, in our view, particularly includes astrocytes [Citation15]. Astrocytes are the most abundant cell type within the central nervous system, and they play essential roles in maintaining normal brain function via contacting, interacting and affecting parenchymal cells [Citation15–Citation17]. Therefore, astrocytes are likely to be essential target for manipulation. Our gene expression microarray revealed that the astrocyte marker GFAP (glial fibrillary acidic protein) was significantly upregulated in the ischemic tissues of transient middle cerebral artery occlusion (tMCAO) stroke model. Whether activated astrocytes perform detrimental or beneficial functions for neuronal survival after an ischemic stroke is still unknown [Citation15]; however, recent studies demonstrate that treatments capable of decreasing infarct size are often accompanied by attenuated astrocyte responses [Citation18,Citation19]. Therefore, careful manipulation of specific pathways that promote the beneficial aspects or reduce the detrimental functions of astrocytes might prove to be valid therapeutic targets for stroke.
Here, using comparative circRNA profiling of ischemic tissues from a tMCAO mouse model and plasma from AIS patients, we identified circRNA HECTD1 (circHECTD1) as an upregulated circRNA. Genome-wide bioinformatic analysis revealed that circHECTD1, which is derived from exons 23 and 24 of the HECTD1 gene, acts as a sponge for MIR142 (microRNA 142). We also found that TIPARP (TCDD inducible poly[ADP-ribose] polymerase), which is predicted to be a target of Mir142, is upregulated in ischemic tissues of the tMCAO mouse model using a gene expression microarray. However, whether the circHECTD1-MIR142-TIPARP axis is involved in cerebral ischemic injury, especially astrocyte activation, remains largely unknown.
Therefore, we hypothesized that upregulated circHECTD1 directly binds to MIR142 and acts as an endogenous MIR142 sponge to inhibit MIR142 activity, which results in astrocyte activation and thus contributes to cerebral infarction. Intervention of circHECTD1 expression significantly inhibited astrocyte activation with amelioration of cerebral infarction. This study indicated that circHECTD1 can be envisioned as a novel biomarker of, and therapeutic target for stroke.
Results
circHECTD1 is upregulated in focal ischemia
Before the experiments, we performed a correlation analysis between cortical cerebral blood flow (CBF) decline (Fig. S1) and neurological deficiency scores after MCAO to control tMCAO efficacy. There was a positive correlation between neurological deficiency scores and the percentage of CBF decline after stroke (Fig. S2). Only animals with a CBF reduction of at least 75% of the baseline level after MCAO were used in the following experiments. To investigate the potential involvement of circRNA in stroke, we performed a circRNA microarray in an established tMCAO mouse model of stroke (GEO accession number: GSE115697). A heat map revealed that after tMCAO, 5 of the 1178 circRNAs analyzed in the circRNA array were upregulated significantly, ≥1.5-fold, in ischemic tissues compared with their levels in sham group (); Table S1). The variation of circRNA expression between the sham and tMCAO samples is shown in scatter and volcano plots in ,). To validate the circRNA microarray results, we first designed head-to-tail junction-specific primers for these circRNAs (labeled with red boxes in )) for use in real-time PCR analysis (); Fig. S3A). Noticeably, only mm9_circ_008488, which was named circHectd1, was highly expressed and dramatically increased in tMCAO group, confirming the increased expression of this circRNA observed in the microarray analysis ()). circHECTD1 is derived from exons 23 and 24 of the HECTD1 gene (Fig. S3B). In addition to the brain, using real-time PCR, we have now verified the expression of circHrctd1 in other organs, such as the heart, liver, spleen, lung and kidney. As shown in Fig. S3C, circHectd1 was highly expressed in the brain and lung compared with its expression in other tissues such as the heart, liver, spleen and kidney. Moreover, circHECTD1 demonstrates high species conservation (Fig. S3D). For example, consistent with the expression of circHectd1 in mouse tissues, circHectd1 was highly expressed in the brain and lung compared with its expression in other tissues such as the heart, liver, spleen and kidney in rats, as shown in Fig. S3E.
Figure 1. circHECTD1 is upregulated in focal ischemia. (A) Microarray heat map representing distinct circRNA expression values in ischemic tissues at 12 h after reperfusion compared with the expression values in the sham tissues (fold change ≥1.5, P < 0.05). Red color scale, higher expression. Green color scale, lower expression. n = 3 samples/group. The statistical significance of the difference was evaluated using Student t tests. (B) A scatter plot assessing the variations in circRNA expression between the sham and tMCAO groups. The values for the X and Y axes are normalized signal values (log2 scaled). The green lines represent fold change lines. circRNAs above the top green line or below the bottom green line change more than 1.5-fold between the 2 groups. (C) Volcano plots assessing the significance of differentially expressed circRNAs. Red points in the plot represent differentially expressed circRNAs that were statistically significant. (D) Divergent primers amplified the circRNAs in cDNA but not gDNA. GAPDH, linear control. M, marker. (E) Validation of the circRNA microarray results. Real-time PCR analysis was used to determine the expression levels of circRNAs in the ischemic cortex from tMCAO and sham mice. n = 8. *P < 0.05 versus the sham group using the Student t test. (F) The circHECTD1 expression levels in plasma from AIS patients (n = 37) and healthy controls (n = 34) measured by real-time PCR. ***P < 0.001 versus the healthy control group using the Student t test.
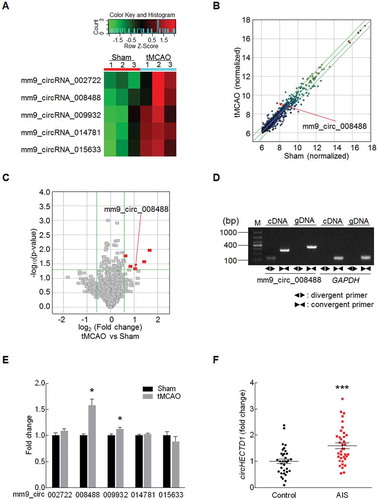
To determine if circRNAs are involved in stroke, we explored the levels of circRNAs in AIS patients. The socio-demographic and clinical characteristics of these subjects are listed in . Plasma samples were collected from AIS patients (age 70.19 ± 1.42 years) who were not undergoing drug therapy. The levels of circHECTD1 in these subjects were significantly increased (P < 0.001, n = 37) compared with those in age- and gender-matched cognitively healthy controls (n = 34) (Figure(f)).
Table 1. Baseline characteristics of the study participants.
Knockdown of circHectd1 expression reduces brain infarction in tMCAO mice
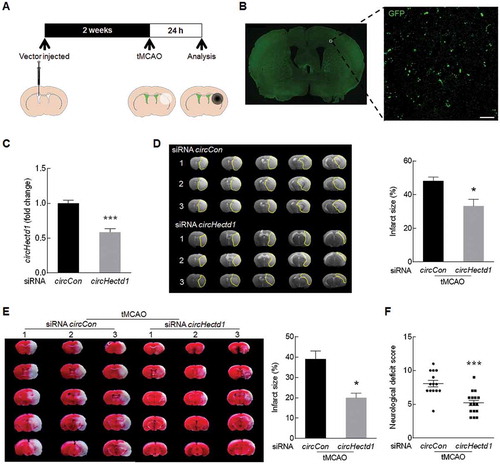
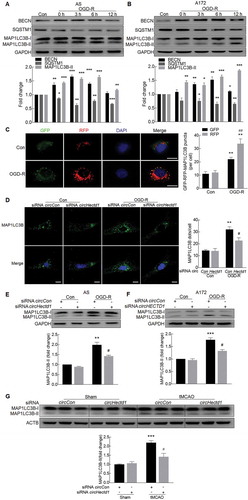
Next, we sought to validate the role of circHectd1 in the pathogenesis of stroke in vivo. Mice were microinjected with either the circCon siRNA lentivirus or the circHectd1 siRNA lentivirus into the lateral ventricle ()). We first examined the efficacy of siRNA circHectd1-GFP lentivirus transduction in vivo. GFP was widely detected in cortex ()), and as expected, decreased circHectd1 expression was observed in the circHectd1 siRNA-injected mice compared with that in the circCon siRNA-injected group ()). Two weeks after the lentivirus microinjection, the mice were processed with tMCAO. After 24 h, infarct size was evaluated by magnetic resonance imaging (MRI) to detect the extent of brain damage. As shown in ), the infarct size was significantly decreased in the mice microinjected with circHectd1 siRNA compared with that in the circCon siRNA-injected mice. To further verify this observation, the brain infarct volume was determined via a 2,3,5-triphenyltetrazolium chloride (TTC) strain assay at 24 h after surgery. The infarct size in the circCon siRNA-injected mice was significantly increased in the tMCAO group, but this increase was inhibited by circHectd1 siRNA injection ()). Consistent with the brain damage findings, the neurological deficiency scores were significantly decreased in circHectd1 siRNA-injected mice compared with those in the circCon siRNA-injected mice after 24 h of reperfusion ()). To further evaluate the role of circHectd1 in long-term recovery, neurological deficiency scores and adhesive removal tests were conducted before and at 3, 7 and 14 days after tMCAO. Microinjection of circHectd1 siRNA resulted in the significant recovery of neurological deficiency (Fig. S4A) and somatosensory functions (Fig. S4B) after tMCAO.
Figure 2. Knockdown of circHectd1 expression reduces brain infarction in tMCAO mice. (A) Illustration of the experimental procedure. (B) The distribution of siRNA circHectd1-GFP lentiviruses in the cortex after lentivirus microinjection. Representative images obtained 2 weeks after the microinjection of circHectd1 siRNA into the lateral ventricle. Green, GFP-circHectd1 siRNA lentivirus. Scale bar: 20 μm. (C) The effect of circHectd1 expression after circHectd1 siRNA lentivirus microinjection. The circHectd1 expression levels at 2 weeks after microinjection were determined by real-time PCR. n = 6 animals/group. ***P < 0.001 versus the circCon siRNA-microinjected group using the Student t test. (D) Representative T2-weighted MRI images of circCon siRNA-microinjected (n = 6) and circHectd1 siRNA-microinjected (n = 11) tMCAO mice. After lentivirus microinjection, the mice were subjected to tMCAO for 60 min and reperfusion for 24 h. *P < 0.05 versus the circCon siRNA-microinjected group using the Student t test. (E) Representative of TTC-stained brain slices. n = 6 animals/group. *P < 0.05 versus the circCon siRNA-microinjected group using the Student t test. (F) Effects of circHectd1 siRNA lentivirus on the neurological deficit score at 24 h after reperfusion. n = 14 animals/circCon siRNA-microinjected group and n = 16 animals/circHectd1 siRNA-microinjected group. ***P < 0.001 versus the circCon siRNA-microinjected group using the Student t test. Con, Control.
Knockdown of circHECTD1 expression inhibits astrocyte activation in vivo
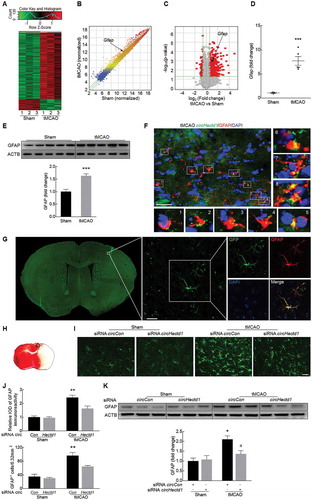
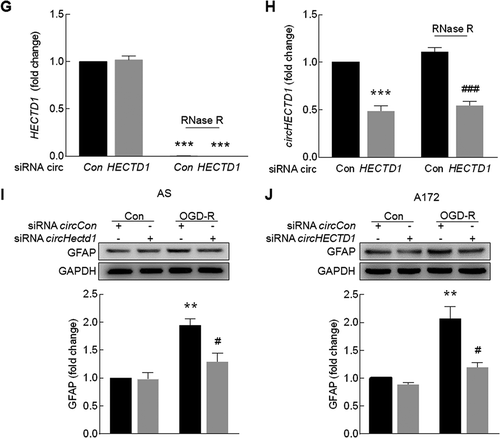
Because circHECTD1 plays a critical role in stroke, we next wanted to examine the mechanisms underlying this process. We first performed an mRNA microarray in a tMCAO mouse model (GEO accession number: GSE98319). A heat map revealed that after tMCAO, 602 of the 19,776 mRNAs analyzed in the mRNA array changed significantly compared with their expression in the sham group ()). The variation of gene expression between the sham and tMCAO groups is shown in scatter and volcano plots in ,). Among these mRNAs, 515 were upregulated by ≥1.5-fold, and 87 were downregulated by ≤0.67-fold in ischemic tissues. We then conducted Gene Ontology (GO) (Fig. S5A, B) and pathway analyses (Fig. S6A and B) for the mRNA-encoding genes to speculate the potential functions of the circRNAs. The most enriched GO categories for the upregulated genes, including ‘response to stress’, ‘immune system process’ and ‘immune response’, have been reported to be involved in the progression of ischemia-reperfusion [Citation20]. To validate the mRNA microarray results, we next examined the expression of GFAP in a tMCAO stroke model. Compared with the sham group, a significant increase in GFAP expression occurred in the ischemic tissue of the tMCAO group, confirming the increased expression of GFAP observed in the microarray analysis (,)). We also found that circHectd1 was colocalized with GFAP in the cortex of tMCAO mice ()). Next, to validate the role of circHectd1 in vivo, circHectd1-siRNA-GFP lentivirus was microinjected into the left lateral ventricle of mice. As shown in ), a certain number of GFAP-positive cells colocalized with GFP. We then immunolabeled brain sections with an astrocyte marker, and the number of activated cells in the cortical penumbra ()) was examined. Mice with tMCAO treatment had increased numbers of reactive astrocytes in the circCon siRNA-injected group, but this effect was significantly ameliorated in the circHectd1 siRNA-injected group (,j)). This finding was further confirmed by western blot analysis in which the expression of GFAP was decreased in the circHectd1 siRNA-injected group compared with that in the circCon siRNA-injected group ()).
Figure 3. Knockdown of circHectd1 expression inhibited astrocyte activation in vivo. (A) A microarray heat map representing distinct mRNA expression values in the ischemic tissues at 12 h after reperfusion compared with those in the sham tissues (fold change ≥1.5, P < 0.05). n = 3 samples/group. The statistical significance of the difference was evaluated using Student t tests. (B) A scatter plot assessing variations in mRNA expression between the sham and tMCAO groups. (C) Volcano plots assessing the significance of differentially expressed mRNAs. (D, E) Real-time PCR (D) and western blot (E) analyses of GFAP expression in the ischemic cortex of tMCAO mice. n = 5 animals/group. ***P < 0.001 versus the sham group using the Student t test. (F) Colocalization of GFAP and circHectd1 in the cortex of tMCAO mice. The areas labeled with the different numbers in the rectangles were enlarged respectively in the bottom or right side of the image. Green, circHectd1; Red, GFAP; Blue, DAPI. Scale bar: 20 μm. (G) Colocalization of GFAP and GFP-circHectd1 siRNA lentiviruses in the cortex of mice. Green, GFP-circHectd1 siRNA lentivirus; Red, GFAP; Blue, DAPI. Scale bar: 20 μm. (H) Astroglial activation is diminished in the cortical penumbra. The rectangle indicates the cortical area used for cell counting. (I) Representative images of astrocytes immunostained for GFAP. Green, GFAP. Scale bar: 50 μm. (J) Quantification of GFAP immunofluorescence intensity and the number of GFAP positive cells using ImageJ software. IOD, integrated optical density. n = 6 animals/group. **P < 0.01 versus the circCon siRNA-microinjected sham group; #P < 0.05 versus the circCon siRNA-microinjected tMCAO group using one-way ANOVA followed by the Holm-Sidak test. (K) Western blot analysis of GFAP after circHectd1 siRNA lentivirus microinjection in tMCAO mice. Mice were treated with tMCAO at 2 weeks after circHectd1 siRNA lentivirus microinjection. The expression of GFAP was determined 24 h after reperfusion. n = 6 animals/group. *P < 0.05 versus the circCon siRNA-microinjected sham group; #P < 0.05 versus the circCon siRNA-microinjected tMCAO group using one-way ANOVA followed by the Holm-Sidak test.
Knockdown of circHECTD1 expression inhibits astrocyte activation in vitro
Because our in vivo study indicated that circHECTD1 was involved in astrocyte activation, we next wanted to examine the role of circHECTD1 in astrocyte activation in vitro. The expression of GFAP was significantly increased in both primary mouse astrocytes ()) and the human glioblastoma A172 cell line ()) treated with oxygen glucose deprivation-reperfusion (OGD-R). Consistent with the in vivo findings, increased expression of circHECTD1 with concomitant decreased expression of HECTD1 in both primary mouse astrocytes ()) and A172 cells ()) was observed. Transduction of astrocytes with circHECTD1 siRNA decreased circHECTD1 expression compared with that observed following transduction with the control siRNA, and the level of HECTD1 was not affected in primary mouse astrocytes ()) and A172 cells ()). To further confirm that transduction with circHECTD1 siRNA only affects circHECTD1, RNA samples were treated with RNase R, which cleaves linear RNAs. As shown in ,h), RNase R treatment resulted in a decreased level of linear HECTD1 mRNA but not circHECTD1. However, transduction with circHECTD1 siRNA decreased the circHECTD1 level in cells treated with RNase R, confirming that siRNA circHECTD1 specifically targets circHECTD1. Transduction of primary mouse astrocytes with circHectd1 ()) or A172 cells with circHECTD1 siRNA ()) significantly inhibited OGD-R-induced astrocyte activation.
Figure 4. Knockdown of circHECTD1 expression inhibited astrocyte activation in vitro. (A, B) Effect of OGD-R on GFAP expression in primary mouse astrocytes (A) and A172 cells (B) as determined by western blot analysis. Cells were treated with OGD for 3 h and reperfusion for 0 h, 3 h, 6 h or 12 h. n = 3. *P < 0.05, **P < 0.01 and ***P < 0.001 versus the control group using one-way ANOVA followed by the Holm-Sidak test. (C, D) Effect of OGD-R on circHECTD1 (left panel) and HECTD1 mRNA (right panel) expression in primary mouse astrocytes (C) and A172 cells (D) as determined by real-time PCR. Cells were treated with OGD for 3 h and reperfusion for 6 h. n = 3. *P < 0.05, **P < 0.01 and ***P < 0.001 versus the control group using the Student t test. (E, F) Effects of circHECTD1 and HECTD1 expression after circHECTD1 siRNA transduction as measured by real-time PCR in primary mouse astrocytes (E) and A172 cells (F). Cells were transduced with circHECTD1 siRNA lentivirus for 48 h and then the expression levels of circHECTD1 and HECTD1 were measured. n = 3. **P < 0.01 and ***P < 0.001 versus the control group using the Student t test. (G, H) Total RNA extracted from the circCon or circHECTD1 siRNA-transfected A172 cells was incubated with or without RNase R followed by real-time PCR. The levels of HECTD1 mRNA (G) and circHECTD1 (H) was detected. ***P < 0.001 versus the no-RNase R-treated group; ###P < 0.001 versus the RNase R-treated siRNA circCon group using one-way ANOVA followed by the Holm-Sidak test. (I, J) circHECTD1 siRNA attenuated OGD-R-induced astrocyte activation in primary mouse astrocytes (I) and A172 cells (J). Cells were transduced with circHECTD1 siRNA lentivirus for 48 h and treated with OGD for 3 h and reperfusion for 6 h. n = 3. **P < 0.01 versus the siRNA circCon group; #P < 0.05 versus the OGD-R-treated siRNA circCon group using one-way ANOVA followed by the Holm-Sidak test. AS, primary mouse astrocytes.
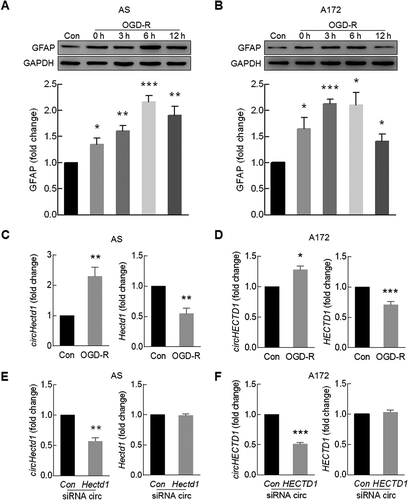
Figure 4. Continued.
circHECTD1 binds MIR142
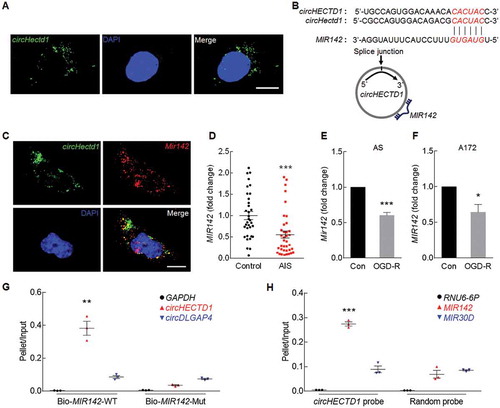
Having determined the essential role of circHECTD1 in the context of stroke, we next wanted to examine the detailed mechanisms underlying circHECTD1-mediated astrocyte activation. We first examined the expression of circHectd1 in primary mouse astrocytes, using fluorescence in situ hybridization (FISH) analysis with a circHectd1-specific probe. As shown in ), circHectd1 was mainly distributed in the cytoplasm, prompting us to investigate whether circHECTD1 functions as a competing endogenous RNA that binds miRNAs via complementary base-pairing. Because circRNAs can act as an endogenous RNA sponge to interact with miRNAs and influence the expression of a target protein, we therefore sought to dissect which miRNA circHECTD1 binds to using the bioinformatics program RNAhybrid. Comparing the sequence of circHECTD1 with MIR142, we found that circHECTD1 contains one target site for MIR142 ()). FISH revealed that circHectd1 and Mir142 colocalize in the cytoplasm ()). Because circHECTD1 was able to bind MIR142, we next investigated MIR142 levels in AIS patients (n = 37), which were significantly decreased compared with those in age- and gender-matched cognitively healthy controls (n = 34) ()). Furthermore, cells exposed to OGD-R exhibited decreased expression levels of MIR142 in primary mouse astrocytes ()) and A172 cells ()). A biotin-coupled MIR142 mimic was applied to test whether MIR142 could affinity-isolate circHECTD1. We observed an enrichment of circHECTD1 in the MIR142-captured fraction compared with that in the fractions that used MIR142 mimics with mutations that disrupt base paring between circHECTD1 and MIR142. However, another circRNA that was not predicted to bind MIR142, circDLGAP4 was not captured by MIR142 ()). This finding was further confirmed by an inverse affinity isolation assay using a biotin-labeled specific circHECTD1 probe as circHECTD1 bound MIR142, but did not bind another miRNA that it was not predicted to bind, such as MIR30D ()).
Figure 5. circHECTD1 binds MIR142. (A) A FISH assay showed the location of circHectd1 in mouse astrocytes. Green, circHectd1; Blue, DAPI. Scale bar: 5 μm. (B) circHECTD1 contains one site complementary to MIR142 as analyzed with RNAhybrid. (C) A FISH assay showed the location of circHectd1 and Mir142 in primary mouse astrocytes. Green, circHectd1; Red, Mir142; Blue, DAPI. Scale bar: 5 μm. (D) The MIR142 expression levels in the serum of AIS patients (n = 37) and healthy controls (n = 34) measured by real-time PCR. ***P < 0.001 versus the healthy control group using the Student t test. (E, F) Effect of OGD-R on MIR142 expression in primary mouse astrocytes (E) and A172 cells (F) as measured by real-time PCR. Cells were treated with OGD for 3 h and reperfusion for 6 h. n = 3. *P < 0.05 and ***P < 0.001 versus the control group using the Student t test. (G) RNA was affinity-isolated by biotinylated WT MIR142 (Bio-MIR142-WT) or its mutant (Bio-MIR142-Mut), and the circHECTD1, circDLGAP4 and GAPDH mRNA levels were quantified by real-time PCR. n = 3. **P < 0.01 versus the bio-MIR142-Mut group using one-way ANOVA followed by the Holm-Sidak test. (H) miRNA was affinity-isolated by the circHECTD1 probe or a random probe, and MIR142, MIR30D and RNU6-6P levels were analyzed by real-time PCR. n = 3. ***P < 0.001 versus the random probe group using one-way ANOVA followed by the Holm-Sidak test.
TIPARP is the downstream target of the circHECTD1-MIR142 axis
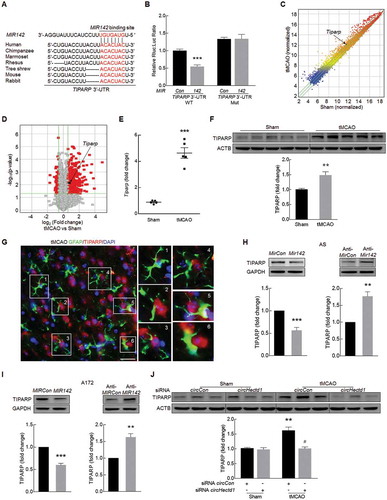
To elucidate the molecular mechanisms by which circHECTD1-MIR142 regulates astrocyte activation in stroke, we next searched for candidate MIR142 target genes predicted by TargetScan. The TIPARP has a conserved MIR142 binding site within its 3ʹ-UTR in most species (). Cotransfection of MIR142 mimic and a pmiR-GLO plasmid with the wild-type TIPARP 3ʹ-UTR resulted in the downregulation of luciferase activity, and this effect was reversed in HEK293T cells transfected with a mutated TIPARP 3ʹ-UTR ()). Interestingly, the same genome wide expression profile indicated that the level of Tiparp was significantly increased in the ischemic tissue of tMCAO mouse model (,)). This finding was further confirmed by the significantly increased expression of TIPARP in the tMCAO group compared with that in the sham group at both the mRNA ()) and protein levels ()). Immunofluorescence staining revealed that GFAP and TIPARP colocalize in the cortex of mice ()). Consistent with this finding, MIR142 decreased TIPARP expression, whereas anti-MIR142 increased its expression in primary mouse astrocytes ()) and A172 cells ()) at the protein level. The in vivo relevance of these findings was further confirmed as microinjection of circHectd1 siRNA lentivirus significantly inhibited the increased expression of TIPARP observed in tMCAO mice ()).
Figure 6. TIPARP is the downstream target of the circHECTD1-MIR142 axis. (A) Putative MIR142 binding sites in TIPARP. The potential complementary residues are shown in red. (B) Relative luciferase activity of wild-type and 3ʹ-UTR mutant constructs of TIPARP cotransfected with MIR142 mimics and miRNA negative control. ***P < 0.001 versus the miRNA negative control group using one-way ANOVA followed by the Holm-Sidak test. (C) A scatter plot assessing the variations in mRNA expression between the sham and tMCAO groups. (D) Volcano plots assessing the significantly differentially expressed mRNAs. (E, F) Real-time PCR (E) and western blot (F) analyses of TIPARP expression in the ischemic cortex of tMCAO mice. n = 5 animals/group. **P < 0.01 and ***P < 0.001 versus the sham group using the Student t test. (G) Colocalization of GFAP and TIPARP in the cortex of mice. Green, GFAP; Red, TIPARP; Blue, DAPI. Scale bar: 10 μm. The areas labeled with the different numbers in the rectangles were enlarged respectively in right side of the image. (H, I) TIPARP expression was evaluated in primary mouse astrocytes (H) and A172 cells (I) transduced with MIRCon-MIR142 and anti-MIRCon-anti-MIR142. n = 4. **P < 0.01 and ***P < 0.001 versus the MIRCon or anti-MIRCon group using the Student t test. (J) Western blot analysis of TIPARP expression after circHectd1 siRNA lentivirus injection in tMCAO mice. n = 6 animals/group. **P < 0.01 versus the circCon siRNA-microinjected sham group; #P < 0.05 versus the circCon siRNA-microinjected tMCAO group using one-way ANOVA followed by the Holm-Sidak test.
The circHECTD1-MIR142 axis promotes astrocyte activation via downstream TIPARP
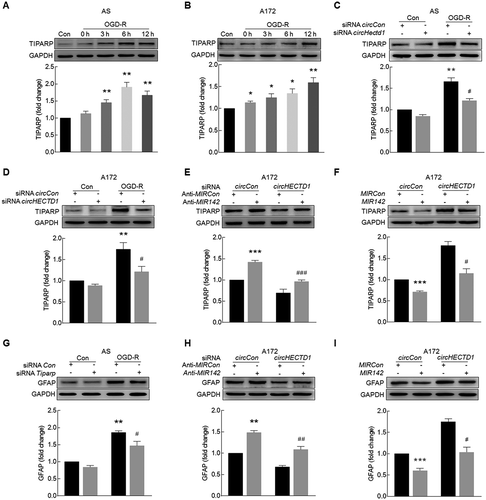
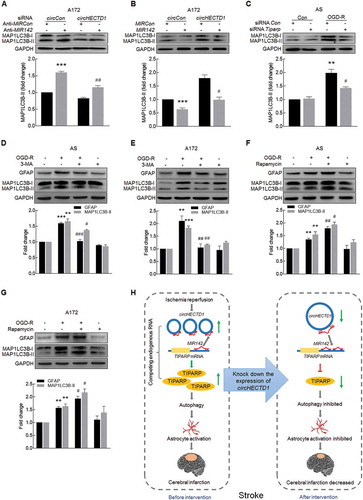
In accordance with this in vivo finding, increased expression of TIPARP was found in both primary mouse astrocytes ()) and A172 cells treated with OGD-R ()). However, transduction of these cells with circHECTD1 siRNA significantly inhibited the increased expression of TIPARP in both primary mouse astrocytes ()) and A172 cells ()) induced by OGD-R. Next, to further verify that MIR142 acts as a mediator of circHECTD1 to control the expression of TIPARP, A172 cells were cotransduced with anti-MIR142 and circHECTD1 siRNA. Knockdown of circHECTD1 attenuated the inductive effects of anti-MIR142 on TIPARP expression in A172 cells ()); however, overexpression of circHECTD1 significantly lessened the decreased expression of TIPARP induced by MIR142 ()). Transduction of cells with circHECTD1 lentivirus, as expected, caused an increase in circHECTD1 expression compared with that in the control without affecting the level of HECTD1 (Fig. S7A and B). The expression of HECTD1 mRNA was further confirmed using another primer designed to target the intron of HECTD1 as ectopic expression of circHECTD1 did not affect host gene HECTD1 expression (Fig. S7C). Consistent with the RNase R experiment above, transduction with circHECTD1 increased the levels of circHECTD1 but not the linear HECTD1 mRNA in cells treated with RNase R, further confirming the circularization of overexpressed circHECTD1 (Fig. S7D and E).
Figure 7. The circHECTD1-MIR142 axis promotes astrocyte activation via its downstream effector TIPARP. (A, B) Effect of OGD-R on TIPARP expression in primary mouse astrocytes (A) and A172 cells (B) as determined by western blot analysis. n = 3. *P < 0.05, and **P < 0.01 versus the control group using one-way ANOVA followed by the Holm-Sidak test. (C, D) circHECTD1 siRNA attenuated OGD-R-induced TIPARP expression in primary mouse astrocytes (C) and A172 cells (D) as determined by western blot analysis. Cells were treated with OGD for 3 h and reperfusion for 6 h. n = 3. **P < 0.01 versus the siRNA circCon group; #P < 0.05 versus the OGD-R-treated siRNA circCon group using one-way ANOVA followed by the Holm-Sidak test. (E) Transduction of A172 cells with circHECTD1 siRNA significantly inhibited anti-MIR142-induced TIPARP expression as determined by western blot analysis. n = 6. ***P < 0.001 versus the circCon siRNA cotransduced with anti-MIRCon group; ###P < 0.001 versus the circCon siRNA cotransduced with anti-MIR142 group using one-way ANOVA followed by the Holm-Sidak test. (F) Transduction of A172 cells with circHECTD1-overexpressed lentivirus significantly increased MIR142-inhibited TIPARP expression as determined by western blot analysis. n = 3. ***P < 0.001 versus the circCon cotransduced with MIRCon group; #P < 0.05 versus the circCon cotransduced with MIR142 group using one-way ANOVA followed by the Holm-Sidak test. (G) Tiparp siRNA attenuated OGD-R-induced GFAP expression in primary mouse astrocytes. Cells were treated with OGD for 3 h and reperfusion for 6 h. n = 3. **P < 0.01 versus the siRNA circCon group; #P < 0.05 versus the OGD-R-treated siRNA circCon group using one-way ANOVA followed by the Holm-Sidak test. (H) Transduction of A172 cells with anti-MIR142 significantly induced circHECTD1 siRNA-inhibited GFAP expression as determined by western blot analysis. n = 3. **P < 0.01 versus the circCon siRNA cotransduced with anti-MIRCon group; ##P < 0.01 versus the circCon siRNA cotransduced with anti-MIR142 group using one-way ANOVA followed by the Holm-Sidak test. (I) Transduction of A172 cells with circHECTD1 overexpressed lentivirus significantly increased MIR142-inhibited GFAP expression as determined by western blot analysis. n = 3. ***P < 0.001 versus the circCon cotransduced with MIRCon group; #P < 0.05 versus the circCon cotransduced with MIR142 group using one-way ANOVA followed by the Holm-Sidak test.
We next sought to examine the role of TIPARP in astrocyte activation using a genetic approach. Transfection of primary mouse astrocytes with Tiparp siRNA efficiently decreased the expression of TIPARP (Fig. S8). Knockdown of Tiparp expression significantly inhibited the astrocyte activation induced by OGD-R ()). Moreover, transduction of A172 cells with anti-MIR142 induced the activation of A172 cells, which was attenuated by siRNA circHECTD1 ()). This finding was further confirmed in A172 cells cotransduced with circHECTD1 and MIR142 ()).
Knockdown of circHECTD1 expression inhibits astrocyte autophagy
Autophagy is a basic cellular process that results in the degradation of defective organelles and misfolded proteins to preserve cellular homeostasis. Mounting evidence indicates that autophagy participates in astrocyte activation [Citation21] and is involved in the pathogenesis of stroke [Citation22–Citation25]. Having determined that circHECTD1 is involved in OGD-R-induced astrocyte activation, we further extended the study and examined whether circHECTD1 regulates astrocyte activation via autophagy. First, we examined the effect of OGD-R on the expression of autophagic proteins in astrocytes. OGD-R treatment of primary mouse astrocytes ()) increased the expression of BECN1/Beclin 1 and induced the production of MAP1LC3B/LC3B-II, which is a cleaved LC3-phosphatidylethanolamine conjugate and a general autophagosomal marker [Citation26]. These findings were further confirmed in A172 cells ()). During the process of autophagy, SQSTM1/p62 acts as a receptor protein that links MAP1LC3B-II with ubiquitin moieties on misfolded proteins. Therefore, autophagy mediates the clearance of SQSTM1 along with ubiquitinated proteins [Citation27]. Consistently, SQSTM1 expression was downregulated by OGD-R treatment in both primary mouse astrocytes ()) and A172 cells ()). Autophagic flux was further monitored in astrocytes that were transduced with a tandem fluorescent mRFP-GFP-MAP1LC3B-adenovirus, a specific marker of autophagosome formation that relies on the differences in GFP and RFP fluorescence under acidic conditions. Colocalization of GFP and RFP signals (yellow dots) indicates a lack of phagophore fusion or of autophagosome and lysosome fusion, whereas RFP-only signals (red dots) indicate the presence of autolysosomes. OGD-R treatment significantly increased the number of yellow dots per cell, with a concomitant increase in RFP-only MAP1LC3B dots in primary mouse microglia transduced with tandem fluorescent-tagged mRFP-GFP-MAP1LC3B ()). Next, we elucidated the effect of circHECTD1 on astrocyte autophagy. Transfection of mouse primary astrocytes with circHectd1 siRNA significantly inhibited OGD-R-induced astrocyte autophagy as determined by immunostaining assay for MAP1LC3B expression ()). This finding was further confirmed by western blot analysis of MAP1LC3B-II in both primary mouse astrocytes ()) and A172 cells ()). Microinjection of circHectd1 siRNA lentivirus significantly inhibited the increased expression of MAP1LC3B-II in tMCAO mice ()).
Figure 8. Knockdown of circHECTD1 expression inhibits astrocyte autophagy. (A, B) Effect of OGD-R on BECN1, SQSTM1 and MAP1LC3B-II expression in primary mouse astrocytes (A) and A172 cells (B) as determined by western blot analysis. n = 3. *P < 0.05, **P < 0.01 and ***P < 0.001 versus the control group using one-way ANOVA followed by the Holm-Sidak test. (C) Primary mouse astrocytes were infected with mRFP-GFP-MAP1LC3B adenovirus and treated with OGD for 3 h and reperfusion for 6 h. Effects of OGD-R on RFP- and GFP- MAP1LC3B puncta (left panel). Scale bar: 5 μm. The numbers of RFP- and GFP- MAP1LC3B puncta per cell were counted (right panel). **P < 0.01 versus the control group; ##P < 0.01 versus the OGD-R group using one-way ANOVA followed by the Holm-Sidak test. (D) MAP1LC3B staining (green) in primary mouse astrocytes transfected with circHectd1 siRNA for 24 h and treated with OGD for 3 h and reperfusion for 6 h. The MAP1LC3B puncta were analyzed by confocal microscopy. Scale bar: 5 μm. **P < 0.01 versus the siRNA circCon group; #P < 0.05 versus the OGD-R-treated siRNA circCon group using one-way ANOVA followed by the Holm-Sidak test. (E, F) Transduction with circHECTD1 siRNA lentivirus attenuated OGD-R-induced MAP1LC3B-II expression in primary mouse astrocytes (E) and A172 cells (F). Cells were treated with OGD for 3 h and reperfusion for 6 h. n = 3. **P < 0.01 and ***P < 0.001 versus the siRNA circCon group; #P < 0.05 versus the OGD-R-treated siRNA circCon group using one-way ANOVA followed by the Holm-Sidak test. (G) circHectd1 siRNA lentivirus injection successfully decreased MAP1LC3B-II expression as determined by western blot analysis. n = 6 animals/group. ***P < 0.001 versus the circCon siRNA-microinjected sham group; #P < 0.05 versus the circCon siRNA-microinjected tMCAO group using one-way ANOVA followed by the Holm-Sidak test.
The circHECTD1-MIR142 axis promotes astrocyte autophagy via downstream TIPARP
Because circHECTD1-MIR142 regulates astrocyte activation, we next examined its role in astrocyte autophagy. Knockdown of circHECTD1 attenuated the inductive effects of anti-MIR142 on autophagy in A172 cells ()). This result was further confirmed in A172 cells cotransduced with circHECTD1 and MIR142. The overexpression of circHECTD1 significantly lessened the decreased expression of MAP1LC3B-II induced by MIR142 ()). Transfection of mouse primary astrocytes with Tiparp siRNA significantly inhibited OGD-R-induced astrocyte autophagy ()).
Figure 9. The circHECTD1-MIR142 axis promotes astrocyte autophagy via its downstream effector TIPARP. (A) Transduction of A172 cells with circHECTD1 siRNA significantly inhibited anti-MIR142-induced MAP1LC3B-II expression as determined by western blot analysis. n = 3. ***P < 0.001 versus the circCon siRNA cotransduced with anti-MIRCon group; ##P < 0.01 versus the circCon siRNA cotransduced with anti-MIR142 group using one-way ANOVA followed by the Holm-Sidak test. (B) Transduction of A172 cells with circHECTD1 overexpressed lentivirus significantly increased MIR142-inhibited MAP1LC3B-II expression as determined by western blot analysis. n = 3. ***P < 0.001 versus the circCon cotransduced with MIRCon group; #P < 0.05 versus the circCon cotransduced with MIR142 group using one-way ANOVA followed by the Holm-Sidak test. (C) Tiparp siRNA attenuated OGD-R-induced MAP1LC3B-II expression in primary mouse astrocytes. Cells were treated with OGD for 3 h and reperfusion for 6 h. n = 3. **P < 0.01 versus the siRNA circCon group; #P < 0.05 versus the OGD-R-treated siRNA circCon group using one-way ANOVA followed by the Holm-Sidak test. (D, E) Pretreatment of cells with an autophagy inhibitor, 3-MA, enhanced OGD-R-induced MAP1LC3B-II and GFAP expression in primary mouse astrocytes (D) and A172 cells (E). Cells were pretreated with 3-MA (2.5 mM) for 1 h and were then treated with OGD for 3 h and reperfusion for 6 h. n = 3. **P < 0.01 and ***P < 0.001 versus the control group; #P < 0.05, ##P < 0.01 and ###P < 0.001 versus the OGD-R group using one-way ANOVA followed by the Holm-Sidak test. (F, G) Pretreatment of cells with an autophagy inducer, rapamycin, attenuated OGD-R-induced MAP1LC3B-II and GFAP expression in primary mouse astrocytes (F) and A172 cells (G). Cells were pretreated with rapamycin (1 μM) for 1 h and were then treated with OGD for 3 h and reperfusion for 6 h. n = 3. **P < 0.01 versus the control group; #P < 0.05 and ##P < 0.01 versus the OGD-R group using one-way ANOVA followed by the Holm-Sidak test. (H) The proposed pathway by which circHECTD1 regulates stroke progression. Upregulated circHECTD1 directly binds to MIR142 and acts as an endogenous MIR142 sponge to inhibit MIR142 activity, which results in astrocyte activation and thus contributes to cerebral infarction as demonstrated in the left dashed frame. Intervention of circHECTD1 expression using siRNA approach will release MIR142 with concomitant downstream downregulation of TIPARP, resulting in the inhibition of astrocyte activation with amelioration of cerebral infarction as demonstrated in the right dashed frame. 3-MA, 3-methyladenine.
We then sought to examine the role of autophagy in astrocyte activation. Both primary mouse astrocytes and A172 cells were treated with an autophagy inhibitor, 3-methyladenine (3-MA), which is a PtdIns3K inhibitor that prevents autophagy at an early stage of autophagosome formation. Pretreatment of cells with 3-MA decreased OGD-R-induced GFAP and MAP1LC3B-II expression in both primary mouse astrocytes ()) and A172 cells ()). In contrast, pretreatment of cells with an autophagy inducer, rapamycin, further enhanced OGD-R-induced astrocyte activation in both primary mouse astrocytes ()) and A172 cells ()). These findings suggest that autophagy enhances astrocyte activation, while the inhibition of autophagy results in the inhibition of astrocyte activation.
Discussion
Even though circRNA profiling has been performed on neurons treated with OGD-R in vitro [Citation28], to the best our knowledge, this report is the first to examine circRNA expression in a tMCAO animal model of stroke using circRNA microarrays. Our study identified 5 previously uncharacterized circRNAs that are significantly upregulated in the ischemic tissues of a tMCAO mouse model. We further characterized circHECTD1, and its upregulation was validated in tMCAO mice as well as in individual AIS patients. Our study elucidated the involvement of the circHECTD1-MIR142-TIPARP axis in ischemic stroke, in particular, its involvement in astrocyte activation and autophagy, which indicates that circHECTD1 can serve as a novel biomarker for and therapeutic target of ischemia-induced astrocyte activation.
circRNAs have important functions in many biological processes. In this study, we found that the level of circHECTD1 was significantly increased in tMCAO mice and in AIS patients, suggesting that a correlation exists between circHECTD1 and the pathogenesis of stroke. We must note that our human study was limited by the availability of only plasma and serum samples, as patient brain tissue samples could not be used. Universality, conserved expression, defined specificity, high stability, and abundance are the main characteristics of circRNAs that render them very attractive diagnostic tools for assessing diseases. Thus far, circRNAs are potential biomarkers for gastric and hepatocellular carcinoma [Citation29,Citation30], and a recent study suggested that circRNAs have the potential to be utilized as biomarkers for neurodegenerative diseases [Citation31]. Here, the findings of our study regarding the expression and function of circHECTD1 indicate that it has the potential to be a promising biomarker for diagnosing AIS and evaluating the degree of damage caused by ischemic insults.
Depending on timing and the stage of brain pathology, astrocyte activity can exacerbate inflammatory reactions and neuronal tissue damage, or it can promote immune suppression and tissue repair [Citation15,Citation32]. Recent studies demonstrate that astrocyte inhibition correlates with decreased infarct size and that treatments capable of decreasing infarct size are often accompanied by attenuated astrocyte responses [Citation18,Citation19]. Consistent with the abovementioned findings, knockdown of circHectd1 significantly decreased infarct size and attenuated neurological deficits with concomitant amelioration of astrocyte activation in tMCAO mice in vivo as well as in astrocytes treated with OGD-R in vitro. To our knowledge, this report is the first to demonstrate that knockdown of circHECTD1 expression inhibits the astrocyte activation induced by OGD-R, revealing a novel function of circHECTD1 in the regulation of astrocyte activation. A better understanding of the role of circHECTD1 in astrocyte activation may facilitate the identification of novel therapeutic targets for the treatment or control of astrocyte activation-related diseases.
circRNAs may act as endogenous sponges that interact with miRNAs to regulate the expression of miRNA target genes. A previous study demonstrated that the circRNA HRCR acts as a MIR223 sponge to regulate cardiac hypertrophy and heart failure [Citation5]. Consistent with previous studies, our current study revealed that circHECTD1 acts a MIR142 sponge to regulate astrocyte activation. The interaction between MIR142 and circHECTD1 was further confirmed by a miRNA affinity-isolation assay. To date, extensive studies have demonstrated that MIR142 is a major regulator of cell fate decisions in the hematopoietic system [Citation33]. Recent evidence has indicated that MIR142 plays pleiotropic roles in embryonic development [Citation34], cancer [Citation35–Citation37], viral infection [Citation38], inflammation and immune tolerance [Citation39,Citation40]. However, few studies have assessed the role of MIR142 in the pathogenesis of stroke. Consistent with previous findings that MIR142 was downregulated in ischemic cardiomyopathy [Citation41], our study identified that MIR142 levels were downregulated in the serum of AIS patients. Although MIR142 has emerged as biomarker for cancer recurrence in patients with early stage lung adenocarcinoma [Citation42] as well as esophageal squamous cell carcinoma [Citation43], to our knowledge, this study is the first to predict that MIR142 is a promising biomarker for the diagnosis of stroke. Consistent with the in vivo findings, our study showed that MIR142 expression was decreased in astrocytes treated with OGD-R and that MIR142 overexpression significantly inhibited OGD-R-induced astrocyte activation. To determine whether circHECTD1-mediated functional effects depend specifically on MIR142, astrocytes were cotransduced with MIR142 and circHECTD1, and overexpression of MIR142 inhibited the astrocyte activation induced by circHECTD1. Consistently, knockdown of circHECTD1 expression significantly counteracted the inductive effect of anti-MIR142 on astrocyte activation. Although the present study has shown that MIR142 is regulated by circHECTD1, our results do not exclude the involvement of other circRNAs or molecules in the regulation of MIR142, either directly or indirectly. Therefore, determining whether miRNAs other than MIR142 bind to circHECTD1 and are involved in astrocyte activation awaits further investigation.
Computational algorithms such as TargetScan have been employed to identify evolutionarily conserved sequences in TIPARP 3ʹ-UTR that are targeted by MIR142. Our current study indicated that the MIR142-TIPARP axis plays a critical role in astrocyte activation in stroke. TIPARP, a critical target induced by constitutively active aryl hydrocarbon receptor signaling [Citation44], is expressed in many different tissues, including the brain, heart, liver, spleen and reproductive organs [Citation45]. The TIPARP contains a CCCH-type zinc finger, a tryptophan-tryptophan-glutamate domain involved in protein-protein interactions and a C-terminal catalytic domain [Citation46]. As a member of the poly (ADP)-ribose polymerase family, the TIPARP transfers mono-ADP-ribose, and not poly-ADP-ribose, to its substrates [Citation47]. ADP-ribosylation is a post-translational modification involved in several biological processes, such as immune cell function, the regulation of transcription, and DNA repair [Citation48]. To date, TIPARP expression is induced by nuclear hormone receptors [Citation49] and platelet-derived growth factor [Citation50] and in response to viral infection [Citation51]. Therefore, the TIPARP likely exerts many effects that are independent of aryl hydrocarbon receptor [Citation52]. In this study, we identified a new regulatory mechanism of TIPARP expression. To our knowledge, this research is the first to demonstrate that TIPARP, as a downstream effector of circHECTD1, regulates the astrocyte activation induced by OGD-R, lending credence to our speculation that the circHECTD1-MIR142-TIPARP axis contributes to astrocyte activation during ischemic stroke. The detailed mechanism by which TIPARP regulates astrocyte autophagy and activation, especially which target transcription factors are involved in this process, needs to be further examined in future studies.
Because autophagy regulates astrocyte activation, we sought to dissect the role of autophagy in astrocyte activation in the context of stroke. Autophagy is a basic cellular process that results in the degradation of defective organelles and misfolded proteins to preserve cellular homeostasis [Citation53,Citation54]. A previous study indicates that autophagy contributes to astrocyte activation, as evidenced by the finding that the autophagy inhibitor 3-MA decreases the astrocyte activation induced by MCAO [Citation21]. Consistent with these findings, our results demonstrated that the autophagy inhibitor 3-MA significantly decreased astrocyte activation, while the autophagy inducer rapamycin had significant inductive effects, suggesting that OGD-R-induced autophagy promotes astrocyte activation. Our in vivo study indicated that knockdown of circHectd1 expression ameliorates neurological deficits in a tMCAO stroke model via inhibition of autophagy, which is consistent with previous studies, which found that microinjection of 3-MA significantly decreased the infarct size induced by MCAO [Citation53,Citation55–Citation58]. However, in contrast to the abovementioned studies (which, as a whole, suggest a deleterious role for autophagy in the tMCAO model), other studies indicated that inhibition of autophagy further worsens neurological deficits in tMCAO stroke model [Citation22,Citation24,Citation25]. In line with these findings, in the context of cerebral ischemia, the role of autophagy is under debate since extensive studies have indicated that autophagy may have different actions in permanent and transient focal ischemia [Citation23].
In terms of the relationship between astrocyte activation and autophagy, our findings seem inconsistent with a previous study that showed the inhibition of slightly but significantly lessened OGD-induced decreases in GFAP expression in the ischemic core at 12 h after permanent MCAO [Citation59]. This discrepancy can be explained by astrocyte death or activation that mainly depends on different stimuli in specific contexts such as astrocyte death induced by OGD for 12 h [Citation60] versus astrocyte activation induced by OGD-R, respectively. If the results obtained in different contexts at first seem contradictory, they in fact simply underscore the complex effects of the multiple pathways activated in astrocytes in the context of neuroinflammation. Therefore, careful manipulation of specific pathways to promote the beneficial aspects and/or to reduce the detrimental effects of reactive astrogliosis might prove to be valid therapeutic targets for a variety of disorders involving a neuroinflammatory component. Further studies are needed to examine the role of specifically regulating astrocyte autophagy in ischemic stroke using genetic approach.
Collectively, our study revealed previously unappreciated regulatory mechanisms of circHECTD1 that regulate astrocyte activation through targeting of the MIR142-TIPARP pathway via the regulation of autophagy ()). Specific blockage of circHECTD1 is predicted to be a potential therapeutic target for the inhibition of astrocyte activation in stroke patients.
Materials and methods
Study approval
The ethics committee at Jiangsu Province Hospital approved this research protocol (approval ID: 2016-SR-235), and the participants or their legally authorized representatives provided written informed consent to participate in the study. All animal procedures were performed in strict accordance with the Animal Research: Reporting of In Vivo Experiments guidelines. The care and use of animals were reviewed and approved by the Institutional Animal Care and Use Committee at the Medical School of Southeast University (approval ID: SYXK-2010.4987).
Human plasma collection
AIS patients were recruited among patients admitted to the Emergency Department at the Affiliated Jiangsu Province Hospital between January 2016 and December 2016. Either an MRI or a computed tomography scan of the brain confirmed the diagnosis of ischemic stroke. Experienced neurologists determined the severity of patients’ neurological deficits using the National Institute of Health Stoke Scale (NIHSS) within 24 h of stroke onset. Patients with intracerebral hemorrhages or unspecified diseases were excluded from the study. The mean time of enrollment blood draw was 16.5 ± 2.6 h post-stroke onset. Non-stroke controls were recruited among patients who underwent an annual medical examination at the hospital. The demographic and clinical characteristics of the 37 patients and 34 non-stroke controls enrolled in the study are provided in .
Animals
Adult male C57BL/6J mice (25.0 to 30.0 g, 6 to 8 weeks old) were purchased from the Model Animal Research Center of Nanjing University (Nanjing, China) and randomly assigned to experimental groups. All animals were housed under a constant temperature and humidity and a 12-h light/12-h dark cycle, with the lights on at 7:00 AM. Food and water were available ad libitum.
Microinjection of circHECTD1 siRNA lentivirus
Eight-week-old C57BL/6J mice were microinjected bilaterally with either the circCon siRNA-GFP lentivirus or the circHectd1 siRNA-GFP lentivirus (1 μl of 1 × 109 viral genomes/µl; GeneChem, contract number: HY20160628FF-YF02) into the left lateral ventricle using the following microinjection coordinates: anteroposterior, −0.3 mm; lateral, 1.0 mm; and ventral, 2.2 mm. To evaluate the effect of siRNA circHectd1 in the tMCAO model, 2 weeks after the microinjection of lentivirus, the mice were divided into 4 groups: siRNA circCon + Sham; siRNA circHectd1 + Sham; siRNA circCon + tMCAO; and siRNA circHectd1 + tMCAO.
tMCAO
tMCAO was processed according to a previous study [Citation61]. In brief, anesthesia was induced with 3% isoflurane mixed with 30% oxygen and 70% nitrous oxide using an anesthetic chamber, and it maintained with 1.5% isoflurane via a facemask. Cannulation of the femoral artery permitted the monitoring of mean arterial blood pressure and arterial blood gases. Rectal temperature was maintained at 37.0 ± 0.5°C during the surgery and the recovery period using a temperature-controlled heating pad. Under a dissecting stereomicroscope, after making a 1-cm long midline skin incision in the neck area, the right common carotid artery was carefully dissected from the surrounding nerves, and a knot was made using 4–0 silk suture. The right external carotid artery (ECA) was exposed and isolated from its small artery branches. The ECA was ligated with 6–0 silk suture approximately 3 mm distal to its origin, and then an arteriotomy was made in the ECA. Next, a silicone-rubber-coated 6–0 nylon filament (Doccol, 602356PK5Re) was inserted into the ECA and advanced over 9–10 mm to the carotid bifurcation along the internal carotid artery and to the origin of the middle cerebral artery. The incision on the neck was sutured, and the mouse was placed in a 35°C nursing box to recover from anesthesia. 1 h after the occlusion, the filament was removed to restore blood flow to the middle cerebral artery territory. In sham-operated mice, the internal carotid artery was surgically prepared for insertion of the filament, but the filament was not inserted. Monitoring of physiological variables was performed in companion cohorts for all of the groups before MCAO and for 60 min after reperfusion. Sham-operated mice served as control.
CBF measurements
Cortical CBF was monitored using a Moor FLPI-2 Full-Field Laser Perfusion Imager, following the manufacturer’s instructions (Moor Instruments, Axminster, UK). Briefly, a charge-coupled device camera was placed above the anesthetized mouse head, and the intact skull surface was illuminated by a laser diode (785 nm) to allow laser penetration through the brain in a diffuse manner. The CBF was measured in both cerebral hemispheres and recorded 15 min before MCAO and throughout the ischemic period until 15 min after the onset of reperfusion. There was no direct sunlight or infrared radiation, and the room temperature was maintained at 26°C. To evaluate CBF changes, the region of interest included the right cortical infarct region, which is posterior to the coronal suture and medial to the linear temporalis. Animals that did not show a CBF reduction of at least 75% of the baseline level or died after ischemic induction (<10%) were excluded from further experimentation [Citation62] because the probability of infarction is greater than 95% if early CBF falls below 25% of the control [Citation63].
In vivo MRI scanning
In vivo MRI was performed using a 7.0-Tesla small animal MRI scanner (Bruker, Ettlingen, Germany, PharmaScan), and T2-weighted images were acquired to calculate the percentage of infarct volume [Citation64,Citation65]. The mice were anesthetized with 2% isoflurane delivered through a nose cone, and their respiratory rate and body temperature were monitored via a physiology monitor. T2-weighted imaging was conducted at 24 h after tMCAO using a 2-dimensional fast-spin echo sequence (2,500/33 msec of repetition time/echo time, 1 average). Twelve axial slices with a slice thickness of 1 mm, a matrix of 256 × 256 and a field of view of 20 × 20 mm were positioned over the brain, excluding the olfactory bulb. The total scan time was 5 min. The percentage of infarct volume was calculated from T2-weighted images. Briefly, the percentage of infarct volume was presented as the lesion volume/the contralateral hemisphere volume covering the whole slices of T2-weighted images. The lesion volume was acquired based on the high signal area of the T2-weighted images from which the artifact of brain edema was subtracted. The infarct volume was traced and calculated using ImageJ software (NIH).
Measurements of neurological deficit and behavioral tests
Neurological deficit was evaluated at 24 h after tMCAO. Neurological deficit assessment was performed by a researcher blinded to the experimental groups. Neurological function measurement was determined using the modified neurological severity scores (mNSS) test [Citation66]. The score was graded on a scale of 0 to 14 (normal score, 0; maximum score, 14). A score of 10 to 14 indicates severe injury; 5 to 9 moderate injury; and 1 to 4 mild injury. For the impairment severity scores, one point is awarded for the inability to perform a test or for the lack of a tested reflex; thus, the higher the score, the severer the injury.
Behavioral tests were performed pre-MCAO and at 3, 7, 14, and 21 days after tMCAO by an independent investigator blinded to the experimental groups. For the adhesive-removal somatosensory test, somatosensory deficit was measured both before and after surgery. All mice were familiarized with the testing environment. In the initial test, 2 small pieces of adhesive-backed paper dots (of equal size, 25 mm2) were used as bilateral tactile stimuli occupying the distal-radial region on the wrist of each forelimb. The mouse was then returned to its cage. The time to remove each stimulus from the forelimbs was recorded for 5 trials per day. Individual trials were separated by at least 5 min. Before surgery, the animals were trained for 3 days. Once the mice were able to remove the dots within 10 seconds, they were subjected to tMCAO.
TTC staining and measurement of cerebral infarction
Infarct volume was evaluated at 24 h after tMCAO. The mice were anesthetized with 1% pentobarbital sodium and perfused with 0.01 M PBS (Gibco, 10,010,001). The mice were then decapitated, and their brains were collected and immediately frozen at −20°C for 6 min. Each brain was coronally sliced into 6 1-mm slices with a brain matrix on ice. The brain slices were incubated in 2% TTC (Sigma-Aldrich, T8877) at 37°C for 10 min and then fixed in 4% paraformaldehyde (PFA) to determine the size and extent of the infarction. The pictures were then analyzed with ImageJ software. To correct for brain swelling, the infarct area was determined by subtracting the area of non-infarcted tissue in the ipsilateral hemisphere from that of the intact contralateral hemisphere. Infarct volume was calculated by integration of the infarct areas for all slices of each brain [Citation67].
Microarray and data analysis
Mice subjected to tMCAO were sacrificed at 12 h after reperfusion. Total RNA was extracted from the ischemic cortex of each mouse using TRIzol reagent according to the manufacturer’s protocol and was quantified using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Sample preparation and microarray hybridization were performed based on the standard protocols of the Arraystar microarray (Arraystar, AS-S-CR-M-1.0 and AS-LNC-M-V3.0). Briefly, for the circRNA microarray, total RNA samples were digested with RNase R (Epicentre, RNR07250) to remove linear RNAs and enrich circular RNAs. Then, the RNAs were amplified and transcribed into fluorescent cRNA utilizing a random priming method (Arraystar Super RNA Labeling Kit; Arraystar). The labeled cRNAs were hybridized onto the Arraystar Mouse circRNA Array (8 x 15 K, Arraystar, AS-S-CR-M-1.0). After washing the slides, the arrays were scanned by the Agilent Scanner G2505C. For the mRNA microarray, total RNA samples were first amplified and transcribed into fluorescent cRNA. Then, the labeled cRNAs were hybridized onto the Arraystar Mouse mRNA Array (8 x 60 K, Arraystar, AS-LNC-M-V3.0). After washing the slides, the arrays were scanned by the Agilent Scanner G2505C.
The acquired array images were analyzed with Agilent Feature Extraction software (version 11.0.1.1, Agilent Technologies). Quantile normalization and subsequent data processing were performed with the Gene Spring GX v11.5.1 software package (Agilent Technologies). Differentially expressed circRNAs and mRNAs between 2 groups were identified through fold-change filtering and Student t tests. Fold-changes greater than 1.5 and P values <0.05 were the criteria used to determine differential expression. Combined analyses were performed using homemade scripts. All of the microarray analysis was performed by KangChen Bio-tech, Shanghai, China. RNAhybrid was used for the prediction of miRNA and circRNA binding sites (https://bibiserv.cebitec.uni-bielefeld.de/rnahybrid) and the prediction of miRNA targets was performed by TargetScan (http://www.targetscan.org/vert_71/).
Real-time PCR
Real-time PCR for mature MIR142 was performed according to our previous studies [Citation68,Citation69] in an Applied Biosystems Real-time PCR System. First, total RNA was extracted using TRIzol reagent (Invitrogen, 15,596,026) and treated with genomic DNA (gDNA) wiper. Then, the RNA was reverse transcribed with a stem-loop RT primer (RiboBio, Guangzhou, China) using a HiScript Q Select RT SuperMix for qPCR Kit (Vazyme, R133-01). Next, the RT products were quantified using AceQ qPCR SYBR Green Master Mix (Vazyme, R141-02). The levels of MIR142 analyzed by real-time PCR were normalized to that of RNU6-6P/RNU6B. Specific primers for mature MIR142 and RNU6-6P were obtained from RiboBio. circRNAs and mRNAs were reverse transcribed using a HiScript Q RT SuperMix for qPCR Kit (Vazyme, R123-01) and quantified using SYBR Green Real-time PCR Master Mix. The primers used to amplify the circRNA and mRNA transcripts are described in Table S2; they were synthesized by Invitrogen. The results were standardized to control values of GAPDH.
Immunostaining and image analysis
Tissue sections were cut into 35-μm slices with a cryostat. The sections were then incubated with 0.3% Triton X-100 (Aladdin, T109027) in PBS for 15 min and blocked with 10% normal goat serum in 0.3% Triton X-100 for 1 h at room temperature. Next, the sections were incubated with a mouse anti-GFAP antibody (1:250; Sigma-Aldrich, G3893), rabbit anti-TIPARP antibody (1:50; Abcam, ab170817) or anti-MAP1LC3B antibody (1:200; Sigma-Aldrich, L8918) overnight at 4°C. On the following day, the sections were washed and incubated with Alexa Fluor 488-conjugated anti-mouse IgG, Alexa Fluor 488-conjugated anti-rabbit IgG or Alexa Fluor 594 goat anti-rabbit IgG (1:250; Invitrogen, A11001/A11008/A11012, respectively) for 1 h. After a final washing step with PBS, the sections were mounted on glass slides, and Prolong gold anti-fade reagent containing DAPI (Southern Biotech, 0100–20) was applied for visualization of nuclei. Immunofluorescence images were captured by microscopy (OLYMPUS, Tokyo, Japan, DP73).
Fish
As in our previous study [Citation69], primary mouse astrocytes cultured on coverslips were fixed with 4% PFA for 20 min and incubated in PBS overnight at 4°C, followed by processing for the detection of circHectd1 or Mir142 expression. The cells were permeabilized with 0.25% Triton X-100 in PBS for 15 min and prehybridized in hybridization buffer (50% formamide, 10 mM Tris-HCl, pH 8.0, 200 μg/ml yeast tRNA [Invitrogen, 15,401,011], 1X Denhardt’s solution [Invitrogen, 750,018], 600 mM NaCl, 0.25% SDS, 1 mM EDTA, 10% dextran sulfate [Sigma-Aldrich, 42,867]) for 1 h at 37°C. Then, the coverslips were heated to 65°C for 5 min in hybridization buffer containing 50 nM of a commercially available biotin-labeled circHectd1 probe (Invitrogen) or 25 nM of a commercially available digoxigenin-labeled Mir142 probe (Invitrogen), and hybridization was allowed to occur at 37°C overnight. The next day, the coverslips were washed 3 times in 2X SSC (10% v/v 20X SSC [Invitrogen, 15,557] in DEPC water [Beyotime, R0021]) and twice in 0.2X SSC at 42°C, blocked with a solution of 1% BSA (Biosharp, BS043D) and 3% normal goat serum (ZSGB-BIO, ZLI-9056) in PBS for 1 h at room temperature and then incubated with a horseradish peroxidase-conjugated anti-digoxigenin antibody (1:200; Roche, 11,207,733,910) and FITC-Streptavidin (1:200; Invitrogen, 434,311) overnight at 4°C. After the coverslips were washed 3 times with TBS (Thermo Scientific, 37,579), signal amplification was carried out using a TSA Cy5 kit (PerkinElmer, NEL745001KT) for 10 min at room temperature. Then, the coverslips were washed twice with PBS and mounted with Prolong gold anti-fade reagent containing DAPI (SouthernBiotech, 010020). The images were captured via microscopy (OLYMPUS, Tokyo, Japan, DP73). The mouse circHectd1 probe sequence, which was biotinylated at the 5ʹ end, was 5ʹ-AAACCATGTTGCAGTTGACCATTGTGGAGCTCGCC-3ʹ. The 5ʹ digoxigenin-labeled Mir142 probe sequence was 5ʹ- TCCATAAAGTAGGAAACACTACA-3ʹ. These probes were all synthesized by Invitrogen.
FISH in combination with immunostaining
As in our previous study [Citation68], brain sections encompassing the entire cortex were cut into 15-μm sections using a cryostat. The sections were fixed in 4% PFA for 20 min and washed with PBS twice. The sections were permeabilized with 0.3% Triton X-100 in PBS for 15 min and prehybridized in hybridization buffer for 1 h at 37°C. Hybridization buffer with 50 nM commercially available biotin-labeled circHectd1 probe was heated to 65°C for 5 min and added dropwise to the sections, which were then allowed to hybridize at 37°C overnight. The next day, the sections were washed 3 times in 2X SSC and twice in 0.2X SSC at 42°C. The sections were blocked with 1% BSA and 3% normal goat serum in PBS for 1 h at room temperature and then incubated with mouse anti-GFAP antibody (1:1,000; Sigma-Aldrich, G3893) overnight at 4°C. On the third day, after the sections were washed 3 times with TBS, they were incubated Alexa Fluor 594 goat anti-mouse IgG (1:250; Invitrogen, A11005) for 1 h at room temperature. The samples were washed twice in PBS and once in DEPC water and were then incubated with Hoechst 33,342 (Invitrogen, H1399) for 10 min at room temperature to visualize the nuclei. Finally, the sections were washed once with DEPC water and mounted with 30% glycerin. Immunofluorescence images were captured using confocal microscopy (ZEISS, Oberkochen, Germany, LSM700).
Cell cultures
Primary mouse astrocytes were obtained from postnatal (P1 to P2) C57BL/6J mice. The mouse brains were removed quickly and placed in ice-cold PBS. After the membranes and large blood vessels were removed, the dissected brain cortices were placed in medium supplemented with PBS. The brain tissues were digested with trypsin-EDTA (Gibco, 25,200,056). Subsequently, the cells were planted on poly-L-lysine precoated cell culture flasks containing DMEM (Corning, 10–013-CVR) supplemented with fetal bovine serum (FBS, 10% v:v; Gibco, 10,099–141) and penicillin/streptomycin (1% v/v, Gibco, 10,378–016). The cultures were maintained in a humidified incubator (37°C, 5% CO2). After 7 to 10 days, the astrocytes were harvested by trypsinization.
The human astrocytoma cell line A172 (ATCC®, RCRL1620TM) was obtained from the China Center for Type Culture Collection, and these cells were routinely maintained in DMEM (with 10% FBS and 1% penicillin/streptomycin) and incubated at 37°C and 5% CO2.
Transduction of astrocytes with lentivirus
Astrocytes were transduced with MIRCon, MIR142, anti-MIRCon, anti-MIR142, circCon, circHECTD1, circCon siRNA or circHECTD1 siRNA lentivirus (HANBIO, Shanghai, China) at a multiplicity of infection of 1 (primary mouse astrocytes) or 10 (A172 cells), followed by gentle swirling, incubation and replacement of fresh feed medium.
OGD-R treatment
OGD-R treatment was performed as described previously [Citation70]. In brief, cells were cultured with deoxygenated DMEM without glucose and FBS (Gibco, 11,966–025) in an incubator (Thermo Fisher Scientific, Waltham, USA) with premixed gas (95% N2 and 5% CO2) for 3 h. The cells were then given normal DMEM containing 10% FBS and placed in a CO2 incubator (95% air and 5% CO2). Cells in the control group were cultured with normal DMEM and 10% FBS for the same incubation times.
Affinity isolation assay with biotinylated miRNA
A total of 2 × 106 HEK293T cells were seeded one day prior to transfection. On the following day, the cells were transfected with 3ʹ-biotinylated MIR142 or MIR142-mutation at a final concentration of 50 nM for 36 h. The cells were then washed with PBS, briefly vortexed, and incubated in lysis buffer (20 mM Tris, pH 7.5, 200 mM NaCl, 2.5 mM MgCl2, 0.05% Igepal [Sigma-Aldrich, 18,896], 60 U/ml Superase-In [Invitrogen, AM2694], 1 mM DTT [Sigma-Aldrich, 43,816], and protease inhibitors [Biotool, B14001]) on ice for 10 min. Lysates were precleared by centrifugation, and 50-μl aliquots of the samples were prepared for input. The remaining lysates were incubated with M-280 Streptavidin magnetic beads (Invitrogen, 11205D). The beads were coated with yeast tRNA in advance to prevent the nonspecific binding of RNA and protein complexes. The beads were incubated at 4°C for 1.5 h and were then washed twice with ice-cold lysis buffer, twice with low-salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.0, 150 mM NaCl), and once with high-salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.0, 500 mM NaCl). The bound RNAs were purified using TRIzol to measure circHECTD1, circDLGAP4 and GAPDH levels. The 3ʹ-biotinylated MIR142-WT sequence was 5ʹ-UGUAGUGUUUCCUACUUUAUGGAUU-3ʹ, and the 3ʹ-biotinylated MIR142- mutation sequence was 5ʹ- UACGGCGGCUCCUACUUUAUGGAUU-3ʹ. The 3ʹ-biotinylated miRNAs were all synthesized by GenePharma (Shanghai, China).
Affinity isolation assay with biotinylated DNA probes
A biotinylated DNA probe complementary to circHECTD1 was synthesized and dissolved in 500 ml of wash and binding buffer (0.5 M NaCl, 20 mM Tris-HCl, pH 7.5, 1 mM EDTA). The probe was incubated with M-280 Streptavidin magnetic beads (Invitrogen, 11205D) at 4°C for 3 h to generate probe-coated magnetic beads. HEK293T cell lysates were incubated with probe-coated beads at 25°C for 1.5 h, and after washing with the wash and binding buffer, the RNA complexes bound to the beads were eluted and extracted for real-time PCR analysis. The sequence of the 3ʹ-biotinylated circHECTD1 probe was 5ʹ-AAACCAAGTTGCAGTTGACCATTGTGTAGTTCTCC-3ʹ, the sequence of the 3ʹ-biotinylated circDLGAP4 probe was 5ʹ-AAAACTAGGCATGATGAACCTTCTTCAGAGAGGTT-3ʹ, and the sequence of the 3ʹ-biotinylated random probe was 5ʹ-AAACAGTACTGGTGTGTAGTACGAGCTGAAGCTAC-3ʹ. These probes were all synthesized by Invitrogen.
Luciferase activity assays
The 3ʹ-UTR of the 708-bp human TIPARP gene containing the putative MIR142 target site was PCR amplified from human genomic DNA using forward (5ʹ-GCGGCTCGAGCCAACCAGAGAACATCAGT-3ʹ) and reverse (5ʹ-AATGCGGCCGCACAAACATTACCAGGAGAG-3ʹ) primers, and the DNA fragment was cloned into the XhoI and NotI sites at the 3ʹ-end of the luc2 gene in the pmiR-RB-REPORTTM vector (RiboBio, Guangzhou, China). For the pmiR-RB-TIPARP-3ʹ-UTR-MIR142-target-mutant vector, the MIR142 target site (ACACTAC) within the TIPARP 3ʹ-UTR was changed to TGTGATG via PCR mutagenesis with the primers TIPARP-MIR142-F (5ʹ-CCTTACTTTGTGATGTTACTTAATAGAAACACA-3ʹ) and TIPARP- MIR142-R (5ʹ-TAAGTAACATCACAAAGTAAGGTACAGAGTCA-3ʹ). Briefly, HEK293T cells were transfected with a MIR142 mimic (RiboBio, Guangzhou, China) and a target plasmid, pmiR-RB-TIPARP-3ʹ-UTR or pmiR-RB-TIPARP-3ʹ-UTR- MIR142-target-mutant, at a molar ratio of 50:1. A miRNA control was used as a negative control. Luciferase activity was determined 24 h post-transfection, and reporter assays were performed following the manufacturer’s protocol (Promega, E2920). Renilla luciferase activity was normalized to firefly luciferase activity and expressed as a percentage of the control.
Western blot analysis
As in our previous studies [Citation71,Citation72], proteins were extracted in RIPA lysis buffer (Beyotime, P0013B), separated on sodium dodecyl sulfate polyacrylamide gels (12% and 15%) and electrophoretically transferred onto polyvinylidene fluoride membranes. The membranes were blocked with 5% non-fat dry milk in Tris-buffered saline with 0.2% Tween-20 (Aladdin, T104863); probed with antibodies recognizing GFAP (1:1,000; Sigma-Alarich, G3893), TIPARP (1:100; Abcam, ab170817), MAP1LC3B (1:1,000; Sigma-Alarich, L7543), BECN (1:1,000; Proteintech, 11,306–1-AP), SQSTM1 (1:1,000; Proteintech, 18,420–1-AP), GAPDH (1:1,000; Santa Cruz Biotechnology, sc-32,233), or ACTB (1:1,000; Bioworld, BS6007M) overnight at 4°C; and then incubated with a horseradish peroxidase-conjugated goat anti-mouse/rabbit IgG secondary antibody (1:2,000; Cell Signaling Technology, 7076P2/7074P2). Signals were detected by chemiluminescence and imaged on a Microchemi 4.2® (DNR, Israel) digital image scanner as described previously [Citation71]. Quantification of the individual protein bands was performed by densitometry using ImageJ software.
Statistics
Statistical analysis was performed using GraphPad Prism 6 Software. All data are presented as the mean ± SEM. Significance was assessed with Student t tests for comparisons of 2 groups or by ANOVA followed by Holm-Sidak tests for 3 or more groups. The appropriate test is indicated in the figure legends. The results were judged to be statistically significant if P < 0.05 by analysis of variance.
Supplemental Material
Download MS Word (33 KB)Acknowledgments
This work was supported by the Major State Basic Research Development Program of China (973 Program) (2013CB733800 and 2013CB733803), the National Natural Science Foundation of China (No. 81673410, No. 81473190 and No. 81603090), the Fundamental Research Funds for the Central Universities and Postgraduate Research & Practice Innovation Program of Jiangsu Province (No. KYZZ16_0127), as well as by grants from the Jiangsu Specially Appointed Professor.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary Material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Jeck WR, Sharpless NE. Detecting and characterizing circular RNAs. Nat Biotechnol. 2014 May;32(5):453–461. PubMed PMID: 24811520; PubMed Central PMCID: PMC4121655.
- Memczak S, Jens M, Elefsinioti A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013 Mar 21;495(7441):333–338. PubMed PMID: 23446348.
- Chen L, Huang C, Wang X, et al. Circular RNAs in eukaryotic cells. Current Genomics. 2015 Oct;16(5):312–318. PubMed PMID: 27047251; PubMed Central PMCID: PMC4763969.
- Zheng Q, Bao C, Guo W, et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun. 2016 Apr 06;7:11215. PubMed PMID: 27050392; PubMed Central PMCID: PMC4823868.
- Wang K, Long B, Liu F, et al. A circular RNA protects the heart from pathological hypertrophy and heart failure by targeting miR-223. Eur Heart J. 2016 Sep 1;37(33):2602–2611. PubMed PMID: 26802132.
- Du WW, Yang W, Chen Y, et al. Foxo3 circular RNA promotes cardiac senescence by modulating multiple factors associated with stress and senescence responses. Eur Heart J. 2017 May 07;38(18):1402–1412. PubMed PMID: 26873092.
- Boeckel JN, Jae N, Heumuller AW, et al. Identification and characterization of hypoxia-regulated endothelial circular RNA. Circ Res. 2015 Oct 23;117(10):884–890. PubMed PMID: 26377962.
- Knowland D, Arac A, Sekiguchi KJ, et al. Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron. 2014 May 07;82(3):603–617. PubMed PMID: 24746419; PubMed Central PMCID: PMC4016169.
- Spescha RD, Shi Y, Wegener S, et al. Deletion of the ageing gene p66(Shc) reduces early stroke size following ischaemia/reperfusion brain injury. Eur Heart J. 2013 Jan;34(2):96–103. PubMed PMID: 23008506.
- Alberts MJ. Stroke treatment with intravenous tissue-type plasminogen activator: more proof that time is brain. Circulation. 2017 Jan 10;135(2):140–142. PubMed PMID: 28069710.
- Snow SJ. Stroke and t-PA–triggering new paradigms of care. N Engl J Med. 2016 Mar 03;374(9):809–811. PubMed PMID: 26962901; PubMed Central PMCID: PMC5331951.
- Denorme F, Langhauser F, Desender L, et al. ADAMTS13-mediated thrombolysis of t-PA-resistant occlusions in ischemic stroke in mice. Blood. 2016 May 12;127(19):2337–2345. PubMed PMID: 26929275.
- Zhou L, Li F, Xu HB, et al. Treatment of cerebral ischemia by disrupting ischemia-induced interaction of nNOS with PSD-95. Nat Med. 2010 Dec;16(12):1439–1443. PubMed PMID: 21102461.
- Rother J. Neuroprotection does not work! Stroke. 2008 Feb;39(2):523–524. PubMed PMID: 18202309.
- Liu Z, Chopp M. Astrocytes, therapeutic targets for neuroprotection and neurorestoration in ischemic stroke. Prog Neurobiol. 2016 Sep;144:103–120. PubMed PMID: 26455456; PubMed Central PMCID: PMC4826643.
- Li M, Li Z, Yao Y, et al. Astrocyte-derived interleukin-15 exacerbates ischemic brain injury via propagation of cellular immunity. Proc Natl Acad Sci U S A. 2017 Jan 17;114(3):E396–E405. PubMed PMID: 27994144; PubMed Central PMCID: PMCPMC5255606.
- Dong YF, Chen ZZ, Zhao Z, et al. Potential role of microRNA-7 in the anti-neuroinflammation effects of nicorandil in astrocytes induced by oxygen-glucose deprivation. J Neuroinflammation. 2016 Mar 09;13(1):60. PubMed PMID: 26961366; PubMed Central PMCID: PMC4785619.
- Wang W, Redecker C, Yu ZY, et al. Rat focal cerebral ischemia induced astrocyte proliferation and delayed neuronal death are attenuated by cyclin-dependent kinase inhibition. J Clinical Neurosci. 2008 Mar;15(3):278–285. PubMed PMID: 18207409.
- Fang SH, Wei EQ, Zhou Y, et al. Increased expression of cysteinyl leukotriene receptor-1 in the brain mediates neuronal damage and astrogliosis after focal cerebral ischemia in rats. Neuroscience. 2006 Jul 07;140(3):969–979. PubMed PMID: 16650938.
- Khan A, Ju F, Xie W, et al. Transcriptomic analysis reveals differential activation of microglial genes after ischemic stroke in mice. Neuroscience. 2017 Apr 21;348:212–227. PubMed PMID: 28223241.
- Xing S, Zhang Y, Li J, et al. Beclin 1 knockdown inhibits autophagic activation and prevents the secondary neurodegenerative damage in the ipsilateral thalamus following focal cerebral infarction. Autophagy. 2012 Jan;8(1):63–76. PubMed PMID: 22108007.
- Su J, Zhang T, Wang K, et al. Autophagy activation contributes to the neuroprotection of remote ischemic perconditioning against focal cerebral ischemia in rats. Neurochem Res. 2014 Nov;39(11):2068–2077. PubMed PMID: 25082119.
- Zhang X, Yan H, Yuan Y, et al. Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy. 2013 Sep;9(9):1321–1333. PubMed PMID: 23800795.
- Yan W, Zhang H, Bai X, et al. Autophagy activation is involved in neuroprotection induced by hyperbaric oxygen preconditioning against focal cerebral ischemia in rats. Brain Research. 2011 Jul 21;1402:109–121. PubMed PMID: 21684529.
- Sheng R, Zhang LS, Han R, et al. Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning. Autophagy. 2010 May;6(4):482–494. PubMed PMID: 20400854.
- Klionsky DJ, Cuervo AM, Seglen PO. Methods for monitoring autophagy from yeast to human. Autophagy. 2007 May-Jun;3(3):181–206. 3678. PubMed PMID: 17224625; eng.
- Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Bio Che. 2007 Aug 17;282(33):24131–24145. PubMed PMID: 17580304.
- Lin SP, Ye S, Long Y, et al. Circular RNA expression alterations are involved in OGD/R-induced neuron injury. Biochem Biophys Res Commun. 2016 Feb 26;471(1):52–56. PubMed PMID: 26845359.
- Chen J, Li Y, Zheng Q, et al. Circular RNA profile identifies circPVT1 as a proliferative factor and prognostic marker in gastric cancer. Cancer Lett. 2017 Mar 01;388:208–219. PubMed PMID: 27986464.
- Qin M, Liu G, Huo X, et al. Hsa_circ_0001649: a circular RNA and potential novel biomarker for hepatocellular carcinoma. Cancer Biomarkers. 2016;16(1):161–169. PubMed PMID: 26600397.
- Kumar L, Shamsuzzama, Haque R, et al. Circular RNAs: the emerging class of non-coding RNAs and their potential role in human neurodegenerative diseases. Mol Neurobiol. 2016 Oct 29 ;54(9):7224–7234. PubMed PMID: 27796758.
- Colombo E, Farina C. Astrocytes: key regulators of neuroinflammation. Trends Immunol. 2016 Sep;37(9):608–620. PubMed PMID: 27443914.
- Nimmo R, Ciau-Uitz A, Ruiz-Herguido C, et al. MiR-142-3p controls the specification of definitive hemangioblasts during ontogeny. Dev Cell. 2013 Aug 12;26(3):237–249. PubMed PMID: 23911199.
- Sladitschek HL, Neveu PA. The bimodally expressed microRNA miR-142 gates exit from pluripotency. Mol Syst Biol. 2015 Dec 21;11(12):850. PubMed PMID: 26690966; PubMed Central PMCID: PMC4704488.
- Isobe T, Hisamori S, Hogan DJ, et al. miR-142 regulates the tumorigenicity of human breast cancer stem cells through the canonical WNT signaling pathway. eLife. 2014 Nov 18:3. PubMed PMID: 25406066; PubMed Central PMCID: PMCPMC4235011.
- Deng B, Zhang Y, Zhang S, et al. MicroRNA-142-3p inhibits cell proliferation and invasion of cervical cancer cells by targeting FZD7. Tumour Bio. 2015 Sep;36(10):8065–8073. 10.1007/s13277-015-3483-2. PubMed PMID: 25976503.
- Chiou GY, Chien CS, Wang ML, et al. Epigenetic regulation of the miR142-3p/interleukin-6 circuit in glioblastoma. Mol Cell. 2013 Dec 12;52(5):693–706. PubMed PMID: 24332177.
- Chaudhuri AD, Yelamanchili SV, Marcondes MC, et al. Up-regulation of microRNA-142 in simian immunodeficiency virus encephalitis leads to repression of sirtuin1. FASEB J. 2013 Sep;27(9):3720–3729. PubMed PMID: 23752207; PubMed Central PMCID: PMC3752547.
- Danger R, Pallier A, Giral M, et al. Upregulation of miR-142-3p in peripheral blood mononuclear cells of operationally tolerant patients with a renal transplant. J Am Soc Nephrol. 2012 Apr;23(4):597–606. PubMed PMID: 22282590; PubMed Central PMCID: PMC3312506.
- Annoni A, Brown BD, Cantore A, et al. In vivo delivery of a microRNA-regulated transgene induces antigen-specific regulatory T cells and promotes immunologic tolerance. Blood. 2009 Dec 10;114(25):5152–5161. PubMed PMID: 19794140; PubMed Central PMCID: PMC2792211.
- Ikeda S, Kong SW, Lu J, et al. Altered microRNA expression in human heart disease. Physiol Genomics. 2007 Nov 14;31(3):367–373. PubMed PMID: 17712037.
- Kaduthanam S, Gade S, Meister M, et al. Serum miR-142-3p is associated with early relapse in operable lung adenocarcinoma patients. Lung Cancer. 2013 May;80(2):223–227. PubMed PMID: 23410826.
- Lin RJ, Xiao DW, Liao LD, et al. MiR-142-3p as a potential prognostic biomarker for esophageal squamous cell carcinoma. J Surg Oncol. 2012 Feb;105(2):175–182. PubMed PMID: 21882196.
- Yamada T, Horimoto H, Kameyama T, et al. Constitutive aryl hydrocarbon receptor signaling constrains type I interferon-mediated antiviral innate defense. Nat Immunol. 2016 Jun;17(6):687–694. PubMed PMID: 27089381.
- Ma Q, Baldwin KT, Renzelli AJ, et al. TCDD-inducible poly(ADP-ribose) polymerase: a novel response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochem Biophys Res Commun. 2001 Nov 30;289(2):499–506. PubMed PMID: 11716501.
- Hottiger MO, Hassa PO, Luscher B, et al. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem Sci. 2010 Apr;35(4):208–219. PubMed PMID: 20106667.
- Vyas S, Matic I, Uchima L, et al. Family-wide analysis of poly(ADP-ribose) polymerase activity. Nat Commun. 2014 Jul 21;5:4426. PubMed PMID: 25043379; PubMed Central PMCID: PMC4123609.
- Kraus WL, Hottiger MO. PARP-1 and gene regulation: progress and puzzles. Mol Aspects Med. 2013 Dec;34(6):1109–1123. PubMed PMID: 23357755.
- Sasse SK, Mailloux CM, Barczak AJ, et al. The glucocorticoid receptor and KLF15 regulate gene expression dynamics and integrate signals through feed-forward circuitry. Mol Cell Biol. 2013 Jun;33(11):2104–2115. PubMed PMID: 23508109; PubMed Central PMCID: PMC3648070.
- Chen WV, Delrow J, Corrin PD, et al. Identification and validation of PDGF transcriptional targets by microarray-coupled gene-trap mutagenesis. Nat Genet. 2004 Mar;36(3):304–312. PubMed PMID: 14981515.
- Atasheva S, Akhrymuk M, Frolova EI, et al. New PARP gene with an anti-alphavirus function. J Virol. 2012 Aug;86(15):8147–8160. PubMed PMID: 22623789; PubMed Central PMCID: PMC3421642.
- Ahmed S, Bott D, Gomez A, et al. Loss of the mono-ADP-ribosyltransferase, Tiparp, increases sensitivity to dioxin-induced steatohepatitis and lethality. J Biol Chem. 2015 Jul 03;290(27):16824–16840. PubMed PMID: 25975270; PubMed Central PMCID: PMC4505429.
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. PubMed PMID: 26799652; PubMed Central PMCID: PMC4835977.
- Kim JY, Zhao H, Martinez J, et al. Noncanonical autophagy promotes the visual cycle. Cell. 2013 Jul 18;154(2):365–376. PubMed PMID: 23870125; PubMed Central PMCID: PMC3744125.
- Zheng C, Han J, Xia W, et al. NAD(+) administration decreases ischemic brain damage partially by blocking autophagy in a mouse model of brain ischemia. Neurosci Lett. 2012 Mar 23;512(2):67–71. PubMed PMID: 22260797.
- Wen YD, Sheng R, Zhang LS, et al. Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy. 2008 Aug;4(6):762–769. PubMed PMID: 18567942.
- Guo Z, Cao G, Yang H, et al. A combination of four active compounds alleviates cerebral ischemia-reperfusion injury in correlation with inhibition of autophagy and modulation of AMPK/mTOR and JNK pathways. J Neurosci Res. 2014 Oct;92(10):1295–1306. PubMed PMID: 24801159.
- Gao L, Jiang T, Guo J, et al. Inhibition of autophagy contributes to ischemic postconditioning-induced neuroprotection against focal cerebral ischemia in rats. PloS One. 2012;7(9):e46092. PubMed PMID: 23029398; PubMed Central PMCID: PMC3461004.
- Qin AP, Liu CF, Qin YY, et al. Autophagy was activated in injured astrocytes and mildly decreased cell survival following glucose and oxygen deprivation and focal cerebral ischemia. Autophagy. 2010 Aug;6(6):738–753. PubMed PMID: 20574158.
- Xu M, Yang L, Hong LZ, et al. Direct protection of neurons and astrocytes by matrine via inhibition of the NF-kappaB signaling pathway contributes to neuroprotection against focal cerebral ischemia. Brain Research. 2012 May 15;1454:48–64. PubMed PMID: 22503072.
- Longa EZ, Weinstein PR, Carlson S, et al. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989 Jan;20(1):84–91. PubMed PMID: 2643202.
- Shi Y, Zhang L, Pu H, et al. Rapid endothelial cytoskeletal reorganization enables early blood-brain barrier disruption and long-term ischaemic reperfusion brain injury. Nat Commun. 2016 Jan 27;7:10523. PubMed PMID: 26813496; PubMed Central PMCID: PMC4737895.
- Heiss WD, Kracht LW, Thiel A, et al. Penumbral probability thresholds of cortical flumazenil binding and blood flow predicting tissue outcome in patients with cerebral ischaemia. Brain. 2001 Jan;124(Pt 1):20–29. PubMed PMID: 11133784.
- Chang D, Wang YC, Bai YY, et al. Role of P38 MAPK on MMP activity in photothrombotic stroke mice as measured using an ultrafast MMP activatable probe. Sci Rep. 2015 Nov 19;5:16951. PubMed PMID: 26581247; PubMed Central PMCID: PMC4652271.
- Bai YY, Gao X, Wang YC, et al. Image-guided pro-angiogenic therapy in diabetic stroke mouse models using a multi-modal nanoprobe. Theranostics. 2014;4(8):787–797. PubMed PMID: 24955140; PubMed Central PMCID: PMC4063977.
- Li Y, Chopp M, Chen J, et al. Intrastriatal transplantation of bone marrow nonhematopoietic cells improves functional recovery after stroke in adult mice. J Cerebral Blood Flow Metabolism. 2000 Sep;20(9):1311–1319. PubMed PMID: 10994853.
- Arumugam TV, Chan SL, Jo DG, et al. Gamma secretase-mediated Notch signaling worsens brain damage and functional outcome in ischemic stroke. Nat Med. 2006 Jun;12(6):621–623. PubMed PMID: 16680150.
- Zhang Y, Shen K, Bai Y, et al. Mir143-BBC3 cascade reduces microglial survival via interplay between apoptosis and autophagy: implications for methamphetamine-mediated neurotoxicity. Autophagy. 2016 Sep;12(9):1538–1559. PubMed PMID: 27464000; PubMed Central PMCID: PMC5082785.
- Yao H, Ma R, Yang L, et al. MiR-9 promotes microglial activation by targeting MCPIP1. Nat Commun. 2014 Jul 14;5:4386. PubMed PMID: 25019481; PubMed Central PMCID: PMC4104446.
- Zhang X, Yuan Y, Jiang L, et al. Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: involvement of PARK2-dependent mitophagy. Autophagy. 2014 Oct 01;10(10):1801–1813. PubMed PMID: 25126734; PubMed Central PMCID: PMC4198364.
- Yao H, Duan M, Buch S. Cocaine-mediated induction of platelet-derived growth factor: implication for increased vascular permeability. Blood. 2011 Feb 24;117(8):2538–2547. PubMed PMID: 21148086; PubMed Central PMCID: PMC3062415.
- Bai Y, Zhang Y, Hua J, et al. Silencing microRNA-143 protects the integrity of the blood-brain barrier: implications for methamphetamine abuse. Sci Rep. 2016 Oct 21;6:35642. PubMed PMID: 27767041; PubMed Central PMCID: PMC5073292.