
ABSTRACT
Deregulation of metabolism during melanoma progression is tightly associated with the genetic and epigenetic alterations of metabolic regulators. Metformin, a macroautophagy/autophagy inducer, has beneficial effects of preventing and treating multiple cancers with an unclear mechanism. Enhanced pseudokinase TRIB3 was reported to link metabolic stressors to melanoma promotion by inhibiting autophagy and ubiquitin-proteasome degradation systems. Here, we discuss our recent findings regarding how metformin reduces TRIB3 expression to restore autophagic flux and suppress melanoma progression in non-diabetic and diabetic mice. We found that overexpression of TRIB3 reverses the metformin-activated autophagic flux, clearance of accumulated tumor-promoting factors and inhibition of tumor progression. Mechanistically, TRIB3 interacts with KAT5 (lysine acetyltransferase 5) and promotes the physical association of KAT5 and SMAD3, which enhances SMAD3 K333 acetylation in a phosphorylation-dependent manner, sustains SMAD3 transcriptional activity and induces TRIB3 expression. Metformin inhibits SMAD3 phosphorylation and impedes the KAT5-SMAD3 interaction, which attenuates the KAT5-mediated K333 acetylation of SMAD3 to suppress SMAD3 transcriptional activity and TRIB3 expression. Our finding defines a molecular mechanism by which metformin targets TRIB3 expression to induce autophagy and protect against melanoma progression.
Metabolic reprogramming is a hallmark of malignant transformation across cancer types and an adaptation to low nutrient and oxygen conditions in the tumor microenvironment that enables cancer cell proliferation. Melanoma, the most aggressive form of skin cancer, develops from melanocytes through the acquisition of multiple genomic alterations including BRAF and NRAS activating mutations and inactivating mutations or deletions in CDKN2A, PTEN and TP53. Recent evidence suggests that these melanoma driver genes control cellular metabolism, and allow melanoma cells to increase glucose consumption and lactic acid production (the Warburg effect) to sustain tumor viability. For instance, BRAFV600E regulates oxidative phosphorylation by suppressing the master regulator of mitochondrial biogenesis, PPARGC1A/PGC1α. Comparatively, BRAFV600E inhibition leads to oxidative addiction through induction of PPARGC1A and increased mitochondrial respiration. Moreover, the combination of BRAF inhibition and modulators of mitochondrial bioenergy metabolism have been suggested to overcome drug resistance in metastatic melanoma. Thus, understanding the molecular and genetic determinants of these metabolic pathways is critical to successfully exploit them for melanoma therapy.
Despite the fact that there is some controversy regarding the roles of autophagy in cancer, the current consensus is that autophagy is a powerful promoter of metabolic homeostasis to prevent tumorigenesis in the early stages of cancer but it promotes the growth of established tumors. For instance, melanoma cells hijack the autophagy machinery to alleviate therapy-induced and metabolic stresses in the tumor microenvironment, thereby promoting resistance to multiple therapies, tumor survival and progression. We have recently determined that metabolic stresses including insulin/IGF elevates expression of TRIB3, a member of the pseudokinase family, which interacts physically with the autophagic receptor SQSTM1 to hinder the binding of SQSTM1 to LC3 and ubiquitinated proteins, leading to SQSTM1 accumulation and inhibition of the clearance of ubiquitinated proteins. Silencing TRIB3 or interrupting the TRIB3-SQSTM1 interaction with an α-helical peptide derived from SQSTM1 attenuates melanoma growth and metastasis through activating autophagic flux and the ubiquitin-proteasome system (UPS). Thus, TRIB3 connects metabolic stressors to tumor progression by inducing a reciprocal antagonism between autophagy and the UPS, 2 main degradation systems. Targeting TRIB3 expression or function is a potential therapeutic strategy for some cancers closely associated with metabolism rewiring. However, directly targeting TRIB3 expression remains a great challenge due to its pseudokinase property.
In a recently published study, we report that metformin, a first-line therapeutic compound for type 2 diabetes (T2D) due to its proven efficacy, high tolerance level and low cost, attenuates melanoma growth and metastasis in non-diabetic C57BL/6 mice and diabetic KK-Ay mice. At the molecular level, metformin maintains effective autophagic flux by downregulating TRIB3 expression and recovering the ‘cargo’ function of SQSTM1, which protects against the accumulation of several tumor-promoting factors in an AMPK-independent manner (). Indeed, rescuing TRIB3 expression abrogates the anti-tumor effects of metformin, even when autophagy initiation signalling is somewhat active. Epidemiological and in vitro studies have supported the rationale that metformin may be used as an adjuvant in chemotherapy for cancer patients. The beneficial effects of metformin as an anti-cancer agent mainly come from its regulating effects on cellular metabolism. Metformin decreases mitochondrial respiration chain activity and ATP production that, in turn, activates AMP-activated protein kinase (AMPK), which regulates energy homeostasis. However, several lines of evidence indicate that AMPK is not required for the beneficial effects of metformin, suggesting additional mechanisms involved in the anti-cancer effects of metformin. Thus, our observations that metformin suppresses melanoma progression by reducing TRIB3 expression and restoring autophagy flux provide new insights into an understanding of the anti-tumor effects of metformin.
Figure 1. Metformin suppresses melanoma progression by reducing SMAD3-mediated TRIB3 expression and restoring autophagy flux. TRIB3 hinders the binding of SQSTM1 to LC3 and ubiquitinated proteins, leading to SQSTM1 accumulation and inhibition of the clearance of ubiquitinated proteins, which promotes tumor growth and metastasis by inhibiting autophagic flux and the ubiquitin-proteasome system (UPS). Metformin attenuates melanoma by reducing TRIB3 expression and activating autophagy; TRIB3 overexpression protects metformin from the activation of autophagic flux and the attenuation of tumor progression. Mechanistically, TRIB3 acts as an adaptor to recruit KAT5 (lysine acetyltransferase 5) to SMAD3 and induce a phosphorylation (P)-dependent K333 acetylation (A) of SMAD3, which sustains transcriptional activity of SMAD3 and subsequently enhances TRIB3 transcription. Metformin suppresses SMAD3 phosphorylation through the following: 1) metformin interacts with the receptor-binding domain of TGFB1 and reduces its binding probability with TGFBR1/TβRI but not TGFBR2/TβRII; 2) metformin suppresses the interaction of TGFBR1 and SMAD3 caused by TGFB1 stimulation. Reduced SMAD3 phosphorylation impedes the KAT5-SMAD3 interaction and protects against the KAT5-mediated K333 acetylation of SMAD3, which reduces SMAD3 transcriptional activity and subsequent TRIB3 expression, thereby restoring autophagy and antagonizing melanoma progression.
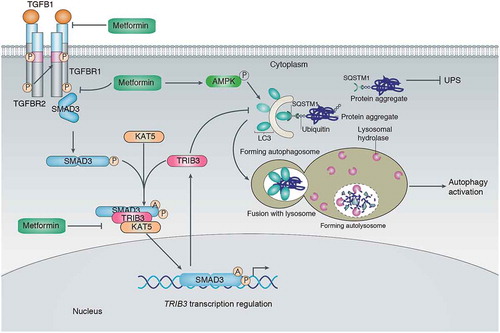
Our previous study revealed that TRIB3 plays a critical role in the promotion of cancer development and progression by interacting with SMAD3 (SMAD family member 3), SQSTM1, or oncoprotein PML-RARA. In this study, we found that TRIB3 maintains the stability of phosphorylated SMAD3, which is crucial for its interaction with KAT5 (lysine acetyltransferase 5), as well as the subsequent acetylation of SMAD3. Moreover, TRIB3 acts as an adaptor to recruit KAT5 and SMAD3 to form a protein complex and enhance KAT5-induced SMAD3 acetylation. The phosphorylation and acetylation of SMAD3 are correlated with SMAD3 transcriptional activity. Metformin inhibits SMAD3 transcriptional activity via suppressing sequential phosphorylation and KAT5-mediated K333 acetylation of SMAD3 to break the positive feedback loop between SMAD3 activity and TRIB3 expression (). Indeed, the correlations among KAT5, SMAD3, and TRIB3 can be observed in human melanoma tissues. Whether the TGFB/TGF-β-SMAD3 pathway is a tumor promoter or suppressor is still a matter of debate due to its differential roles in the early and late stages of carcinogenesis. Notably, KAT5 may act either as a tumour suppressor by acetylating TP53 or ABL to increase apoptosis in many cancer types, including melanoma, or as a tumor enhancer by increasing MYC, NF-κB and AR (androgen receptor) transcriptional activation. Further work will be required to determine how KAT5 acts as a double-edged sword in cancer, similar to the way that TGFB-SMAD3 signalling does.