
ABSTRACT
Macroautophagy/autophagy, a defense mechanism against aberrant stresses, in neurons counteracts aggregate-prone misfolded protein toxicity. Autophagy induction might be beneficial in neurodegenerative diseases (NDs). The natural compound trehalose promotes autophagy via TFEB (transcription factor EB), ameliorating disease phenotype in multiple ND models, but its mechanism is still obscure. We demonstrated that trehalose regulates autophagy by inducing rapid and transient lysosomal enlargement and membrane permeabilization (LMP). This effect correlated with the calcium-dependent phosphatase PPP3/calcineurin activation, TFEB dephosphorylation and nuclear translocation. Trehalose upregulated genes for the TFEB target and regulator Ppargc1a, lysosomal hydrolases and membrane proteins (Ctsb, Gla, Lamp2a, Mcoln1, Tpp1) and several autophagy-related components (Becn1, Atg10, Atg12, Sqstm1/p62, Map1lc3b, Hspb8 and Bag3) mostly in a PPP3- and TFEB-dependent manner. TFEB silencing counteracted the trehalose pro-degradative activity on misfolded protein causative of motoneuron diseases. Similar effects were exerted by trehalase-resistant trehalose analogs, melibiose and lactulose. Thus, limited lysosomal damage might induce autophagy, perhaps as a compensatory mechanism, a process that is beneficial to counteract neurodegeneration.
Abbreviations: ALS: amyotrophic lateral sclerosis; AR: androgen receptor; ATG: autophagy related; AV: autophagic vacuole; BAG3: BCL2-associated athanogene 3; BECN1: beclin 1, autophagy related; CASA: chaperone-assisted selective autophagy; CTSB: cathepsin b; DAPI: 4ʹ,6-diamidino-2-phenylindole; DMEM: Dulbecco’s modified Eagle’s medium; EGFP: enhanced green fluorescent protein; fALS, familial amyotrophic lateral sclerosis; FRA: filter retardation assay; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; GLA: galactosidase, alpha; HD: Huntington disease; hIPSCs: human induced pluripotent stem cells; HSPA8: heat shock protein A8; HSPB8: heat shock protein B8; IF: immunofluorescence analysis; LAMP1: lysosomal-associated membrane protein 1; LAMP2A: lysosomal-associated membrane protein 2A; LGALS3: lectin, galactose binding, soluble 3; LLOMe: L-leucyl-L-leucine methyl ester; LMP: lysosomal membrane permeabilization; Lys: lysosomes; MAP1LC3B: microtubule-associated protein 1 light chain 3 beta; MCOLN1: mucolipin 1; mRNA: messenger RNA; MTOR: mechanistic target of rapamycin kinase; NDs: neurodegenerative diseases; NSC34: neuroblastoma x spinal cord 34; PBS: phosphate-buffered saline; PD: Parkinson disease; polyQ: polyglutamine; PPARGC1A: peroxisome proliferative activated receptor, gamma, coactivator 1 alpha; PPP3CB: protein phosphatase 3, catalytic subunit, beta isoform; RT-qPCR: real-time quantitative polymerase chain reaction; SBMA: spinal and bulbar muscular atrophy; SCAs: spinocerebellar ataxias; siRNA: small interfering RNA; SLC2A8: solute carrier family 2, (facilitated glucose transporter), member 8; smNPCs: small molecules neural progenitors cells; SOD1: superoxide dismutase 1; SQSTM1/p62: sequestosome 1; STED: stimulated emission depletion; STUB1: STIP1 homology and U-box containing protein 1; TARDBP/TDP-43: TAR DNA binding protein; TFEB: transcription factor EB; TPP1: tripeptidyl peptidase I; TREH: trehalase (brush-border membrane glycoprotein); WB: western blotting; ZKSCAN3: zinc finger with KRAB and SCAN domains 3
Introduction
Macroautophagy (autophagy hereafter) is a well-conserved degradative pathway, which is part of intracellular quality control systems in almost all eukaryotic cell types. The major physiological role of autophagy is to maintain a regular energy supply by recycling macromolecules, especially during starvation or under several types of cell stresses [Citation1]. Autophagy also assures the renewal of intracellular molecular and macromolecular components eliminating damaged proteins and organelles. In addition, autophagy acts against infection by destroying invading intracellular bacteria and viruses [Citation1]. Mutations in genes correlated to autophagy are associated with different pathological conditions, such as cancer and neurodegenerative diseases (NDs) [Citation2]. In these 2 classes of human diseases, autophagy is activated and/or blocked in a way that may differentially correlate with disease progression [Citation2]. In particular, in NDs associated with the accumulation of misfolded proteins (e.g., Parkinson disease [PD], Huntington disease [HD], amyotrophic lateral sclerosis [ALS], spinal and bulbar muscular atrophy [SBMA], several types of spinocerebellar ataxias), activation of autophagy represents one of the most efficient arms of the protein quality control system, which removes neurotoxic protein species [Citation3]. An insufficient or blocked autophagic flux generally correlates with misfolded protein accumulation into intracellular inclusions in different neuronal compartments (nucleus, cytoplasm, axons, mitochondria, endoplasmic reticulum, etc.) [Citation4–Citation6]. Because of that, several investigators have attempted to pharmacologically potentiate the autophagic flux in neuronal cells (or other cell types involved in disease, such as glial or muscle cells) to enhance the clearance of neurotoxic misfolded proteins.
Interestingly, among autophagy inducers, the natural disaccharide trehalose, widely used as nutraceutical, preservative, and humectant, exerts a potent pro-autophagic activity both in vitro and in vivo [Citation7–Citation9]. Notably, trehalose enhances the clearance of several different mutant misfolded proteins responsible for neuronal death, in different neuronal models of adult onset NDs [Citation9–14]. In vivo, trehalose promotes survival of several mouse models of ND (human tauopathy, HD, PD, ALS, oculopharyngeal muscular dystrophy) [Citation13,Citation15–Citation26], either/both by delaying onset or/and by slowing down progression of the diseases, generally with an amelioration of the symptoms associated with the disease. Despite these very promising observations, the mechanism by which trehalose activates and increases the autophagic flux is still obscure. Trehalose induces de novo expression of 2 autophagic proteins, SQSTM1/p62 and MAP1LC3B, and activates the conversion of MAP1LC3B-I to its autophagosome-associated lipidated form LC3-II [Citation9–Citation11,Citation13,Citation27–Citation29]. Trehalose also induces expression of a potent facilitator of the autophagic flux, the small heat shock protein HSPB8 [Citation10]. However, pro-autophagic activity of trehalose does not appear to depend on the MTOR (mechanistic target of rapamycin kinase) pathway, an important sensor of the cell nutrient status [Citation9,Citation16,Citation24,Citation28,Citation30].
It has been suggested that trehalose activity could be mediated by the inhibition of a glucose transporter [Citation31,Citation32], SLC2A8/GLUT8 (solute carrier family 2, [facilitated glucose transporter], member 8), thus preventing glucose uptake into cells [Citation32]. The reduction of glucose uptake could produce an apparent ‘starvation,’ which activates autophagy [Citation32,Citation33]. However, it has been shown that trehalose itself needs to be internalized in cells to activate autophagy, because deletion of the gene encoding SLC2A8, which also acts as a trehalose importer, blocks the ability of trehalose to induce autophagy [Citation33].
Interestingly, trehalose induces nuclear translocation of TFEB (transcription factor EB) [Citation30,Citation34] a master regulator of the expression of many autophago-lysosomal components [Citation35–Citation40]. Several kinases have been suggested to mediate this activity. For example, by regulating FOXO3/FOXO3A phosphorylation, trehalose could activate the adenosine 5ʹ-monophosphate-activated protein kinase, thereby enhancing the activity of CARM1 (coactivator-associated arginine methyltransferase 1), a TFEB co-activator [Citation32]; alternatively, trehalose could inhibit AKT, a kinase which retains TFEB in its inactive phosphorylated status in the cytoplasm [Citation30].
Here, we show that trehalose can regulate autophagy by a mechanism involving changes in lysosomes with their rapid enlargement and transient permeabilization upon treatment, as we observed by super-resolution microscopy and electron microscopy. We find that such lysosomal modifications correlate with the activation of PPP3CB/calcineurin (protein phosphatase 3, catalytic subunit, beta isoform), likely via lysosomal calcium release. Activated PPP3CB targets and specifically dephosphorylates TFEB, inducing translocation from the cytoplasm to the nucleus and conferring transcriptional competence. Thus, transient lysosomal damage could directly promote TFEB activation, ultimately activating autophagy upon trehalose stimulation.
Results
Trehalose induces TFEB nuclear translocation and activation of autophagy-linked genes in immortalized motoneurons
Trehalose treatment counteracts misfolded protein toxicity in models of ND, including ALS and SBMA, in which motoneurons die. Thus, we initially evaluated whether trehalose induces TFEB nuclear translocation in NSC34, an immortalized motoneuronal cell line. Confocal immunofluorescence (IF) microscopy showed that in untreated conditions endogenous TFEB was confined to the cell cytoplasm (, quantification in 1B). After 2 h of trehalose treatment, a different pattern of distribution was appreciable, with TFEB appearing in the nucleus. However, only at 18–24 h of trehalose treatment a clear nuclear staining become detectable, with a massive nuclear compartmentalization at 48 h.
Figure 1. Trehalose activates TFEB nuclear translocation and induces protein quality control genes. (a-m) NSC34 cells were treated with 100 mM trehalose or glucose (as control) for different times. (a) IF analysis performed with anti-TFEB antibody (green), nuclei were stained with DAPI (blue) (63X magnification). Scale bar: 10 μm. (b) The bar graph represents the quantification of TFEB nuclear intensity; the fields were randomly selected and at least 100 cells for each sample were analyzed (n = 3) (*p < 0.05, ** p < 0.005, *** p < 0.001, one-way ANOVA with Tukey’s test). (c) WB analysis of cytoplasmic (C) and nuclear extracts (N). GAPDH and histone H3 were used as loading controls for cytoplasmic and nuclear fractions, respectively. (d-m) RT-qPCR analyses. The relative fold difference in mRNA expression was determined using t = 0 as internal control. Data are means ± SD of 4 independent samples. RT-qPCR on the following mRNA: Tfeb (d); Zkscan3 (e); Sqstm1/p62 (f); Map1lc3b (g); Atg10 (h); Atg12 (i); Hspb8 (j); Bag3 (k); Becn1 (l); Ppargc1A (m). Bar graphs represent the relative fold induction of these genes (*p < 0.05, ** p < 0.005, *** p < 0.001, one-way ANOVA with Tukey’s test).
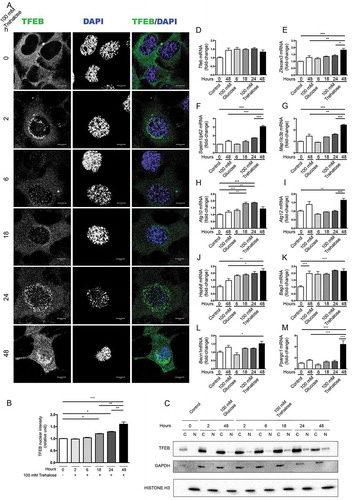
To confirm trehalose-induced nuclear relocalization of TFEB, we performed a fractionation experiment ()). We found that the nuclear TFEB levels started to rise 18 h after trehalose treatment and gradually increased at later stages. Conversely, cytoplasmic TFEB protein levels were sharply decreased after 48 h of trehalose treatment. These data confirm previous findings that trehalose may induce TFEB activation and suggest that the TFEB-mediated induction of autophagy might be a late event of trehalose treatment.
We next tested whether trehalose treatment could lead to transcriptional activation of TFEB targets. To this end, we analyzed whether TFEB translocation correlated with an enhanced transcription of selected pro-autophagic genes. panels d-m show the RT-qPCR performed on RNAs obtained from trehalose-treated or control (glucose treated) NSC34 cells at different times after treatments. Trehalose was unable to modify the expression levels of Tfeb () whereas it specifically enhanced the expression of genes coding for the autophagy inducer BECN1 (), SQSTM1/p62 (sequestosome 1) (), which is an autophagy receptor, and its interactor MAP1LC3B (microtubule-associated protein 1 light chain 3 beta) (), which is required for autophagosome formation. The enhanced expression of these genes was present after 48 h of treatment, in line with the observed TFEB nuclear translocation (). Trehalose also induced the de novo expression of 2 autophagy related (Atg) genes, coding respectively for the E2-like enzyme ATG10 () and for ATG12 which, in conjunction with ATG5, acts as an E3-like enzyme required for the MAP1LC3B lipidation and activation (). However, these 2 Atg genes differentially respond to TFEB activation, because Atg10 was significantly induced by trehalose at 18–24 h, whereas Atg12 was induced at a later time point (48 h after trehalose exposure). After 48 h of trehalose treatment, we found a robust increase of the expression levels of an additional TFEB responder, Ppargc1A (peroxisome proliferative activated receptor, gamma, coactivator 1 alpha), that transcriptionally regulates genes involved in nutrient metabolism and acts as a master regulator of mitochondrial biogenesis [Citation41–Citation43] (). Surprisingly, trehalose treatment also significantly induced the expression of Zkscan3 (zinc finger with KRAB and SCAN domains 3) (), coding for a master repressor of autophagy, which acts as a physiological antagonist of TFEB [Citation44]. Changes in Zkscan3 expression were detectable after 48 h of trehalose treatment, suggesting that its expression might be an attempt to limit the autophagy induction associated with TFEB activation.
In several NDs, clearance of misfolded proteins by autophagy is assisted by the chaperone-assisted selective autophagy (CASA) complex [Citation45–Citation48], which depends on HSPB8. HSPB8 is a chaperone induced by trehalose, which facilitates autophagic degradation of misfolded proteins acting both as a limiting factor for the CASA complex and a regulator of the proteasome and autophagic routing systems [Citation10,Citation45,Citation49–Citation54]. In the CASA complex, HSPB8 (heat shock protein 8) binds BAG3 (BCL2-associated athanogene 3) and the HSPA8 (heat shock protein 8)-STUB1 (STIP1 homology and U-box containing protein 1) complex allowing STUB1-mediated ubiquitination of target misfolded substrates followed by SQSTM1/p62 recognition and insertion into the autophagy pathway. We thus analyzed whether expression of HSPB8 and BAG3 was modified by trehalose treatment (). Hspb8 expression was found induced by trehalose treatment, whereas Bag3 was induced by both trehalose and glucose treatment, possibly as a general response to osmotic variations. Hspb8, Sqstm1/p62, Map1Lc3b and Atg12 inductions were maximal at 48 h after trehalose exposure, suggesting a cooperative activity between these proteins. Overall, these data suggest that trehalose acts by inducing genes that are typically controlled by TFEB.
TFEB downregulation prevents trehalose-mediated autophagy induction
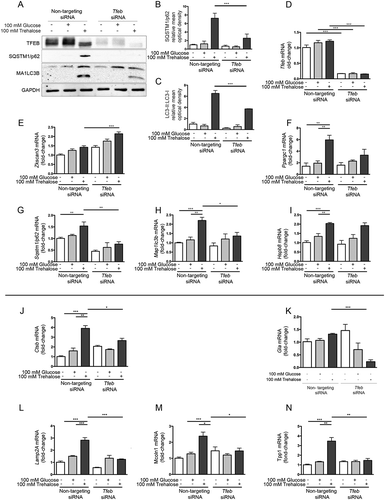
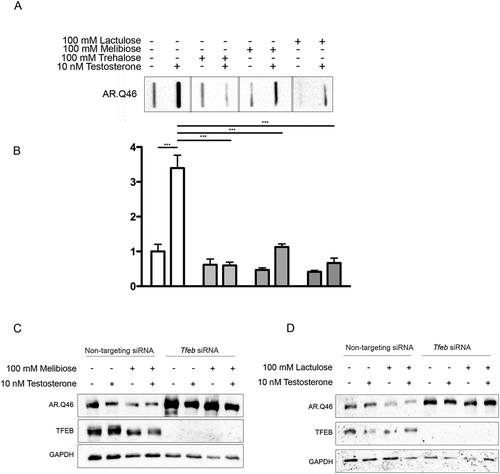
While trehalose activates expression of autophagy genes in a time window compatible with nuclear translocation of endogenous TFEB, it is unclear if these 2 effects are directly related. To assess this, we next determined whether trehalose requires TFEB to exert its action. Tfeb siRNA efficiently downregulated TFEB protein () and mRNA () levels, while a non-targeting siRNA was inactive. Interestingly, in line with their transcriptional activation (), trehalose treatment strongly induced SQSTM1/p62 and MAP1LC3B-I (as well as its conversion to the MAP1LC3B-II lipidated form, required to sustain autophagy flux); this effect of trehalose was severely blunted when TFEB was downregulated with its specific siRNA (, quantifications 2b and 2c). The involvement of TFEB in mediating the action of trehalose on these 2 genes was also confirmed by RT-qPCR analysis (), which revealed that TFEB downregulation resulted in a significant decrease of both Sqstm1/p62 and Map1Lc3b as well as of Ppargc1A expression (). Notably, TFEB downregulation did not affect trehalose-induced expression of the Zkscan3 gene, suggesting that expression of this autophagy repressor is independent of a transcriptional regulation mediated by TFEB (). Similarly, TFEB downregulation did not modify Hspb8 expression (). We also analyzed other TFEB regu–lated genes involved in lysosomal dynamics: those encoding lysosomal enzymes, Ctsb (cathepsin b) () Gla (galactosidase, alpha) (), and Tpp1 (tripeptidyl peptidase 1) (), and lysosomal membrane proteins, Lamp2a (lysosomal-associated membrane protein 2A) () and the calcium channel Mcoln1 (mucolipin 1) (). Trehalose treatment induced a robust increase in the expression of most of these lysosomal genes, except for Gla, which was marginally induced after trehalose treatment. In line with our previous observation, also in this case, TFEB downregulation markedly reduced the trehalose-induced expression of these genes. These results indicate that, upon trehalose treatment, TFEB is crucial for induction of autophagy and lysosomal functionality, but dispensable for expression of Zkscan3 and Hspb8.
Figure 2. TFEB silencing counteracts trehalose-induced expression of autophagic genes. (a-n) NSC34 cells were transfected with Tfeb or non-targeting (as control) siRNAs and treated with 100 mM glucose or trehalose. (a) WB analysis was performed, and the bar graphs (b-c) represent the mean relative optical density of SQSTM1/p62 and MAP1LC3B-II:MAP1LC3B-I protein expression levels, respectively, performed with n = 3 independent samples. LC3-II:LC3-I ratio was calculated by densitometric analysis of both bands. (*** p < 0.001, two-way ANOVA with Tukey’s test.) (d-n) RT-qPCR for the following mRNA: Tfeb (d); Zkscan3 (e); Ppargc1a (f); Sqstm1/p62 (g); Lc3 (h); HspB8 (i); Ctsb (j); Gla (k); Lamp2A (l); Mcoln1 (m); Tpp1 (n); the bar graphs represent the relative fold induction of these genes normalized with Rplp0 mRNA levels. Data are means ± SD of 4 independent samples (** p < 0.005, *** p < 0.001, two-way ANOVA with Bonferroni’s test).
Trehalose promotes clearance of neurotoxic misfolded proteins in a TFEB-dependent manner
To verify whether the action of trehalose specifically on polyQ-containing proteins in NDs is mediated by TFEB, we made use of cells expressing a mutant AR (androgen receptor), which contains an elongated polyglutamine tract (ARpolyQ). ARpolyQ is responsible for spinal and bulbar muscular atrophy, and others and we have previously demonstrated that its cellular aggregation is cleared following trehalose treatment [Citation10,Citation27,Citation29]. One advantage of the ARpolyQ cell model is that aggregation and associated neurotoxicity is triggered by the ligand testosterone [Citation10,Citation27,Citation55–Citation60]. Consistent with this, using a filter retardation assay (FRA) and western blotting (WB) [Citation10,Citation49,Citation58,Citation61,Citation62] we found that aggregation of AR.Q46 (a form of ARpolyQ containing 46 polyglutamines) is triggered by 10 nM testosterone, and that trehalose reduces the monomeric AR.Q46 (), a process that we already demonstrated to be mediated by autophagy activation [Citation10,Citation27], and fully reverts AR.Q46 aggregation (, quantification in ). Importantly, TFEB downregulation completely abolished the prodegradative activity of trehalose on testosterone-induced monomeric and aggregated AR.Q46 forms (), proving that trehalose enhances ARpolyQ clearance via the TFEB pathway. These data have been confirmed also in iPSCs derived from SBMA patients and differentiated to motoneuronal-like cells. In fact, we found that trehalose reduced the overall levels of AR.Q51 in a WB assay () increasing the MAP1LC3B-I to MAP1LC3B-II conversion.
Figure 3. TFEB silencing counteracts trehalose-induced pro-degradative effects. (a-c) NSC34 cells were transfected with Tfeb siRNA or non-targeting siRNAs, and with AR.Q46, untreated or treated with 100 mM trehalose, in absence or in presence of 10 nM testosterone for 48 h. (a) WB analysis and (b) FRA were performed. (c) The bar graph represents the mean relative optical density of FRA ± SD for n = 3 independent samples (*** p < 0.001, two-way ANOVA with Bonferroni’s test). (d) WB analysis on iPSCs derived from SBMA patient (Q51) and differentiated to motoneuronal-like cells for 10 days in absence or in presence of 10 nM testosterone, and treated with 100 mM trehalose for the last 48 h. WB blot analysis was performed. Histone H3 was used for loading control. (e-g) NSC34 cells were transfected with Tfeb siRNA or non-targeting siRNA, and with GFP-TARDBP-25 for 48 h, untreated or treated with 100 mM trehalose for 48 h (e) WB analysis and (f) FRA were performed. (g)The bar graph represents the mean relative optical density of FRA ± SD for n = 3 independent samples (*p < 0.05, two-way ANOVA with Bonferroni’s test). (h-m) NSC34 cells were transfected with Tfeb siRNA or non-targeting siRNA, and with mutant GFP-SOD1 (SOD1A4V and G93A), for 48 h, untreated or treated with 100 mM trehalose for 48 h. WB analysis (h, k) and FRA were performed (i,l). (j,m) The bar graphs represent the mean relative optical density of FRA ± SD for n = 3 independent samples (*p < 0.05, two-way ANOVA with Bonferroni’s test). For WB experiments, GAPDH was used as an internal loading control.
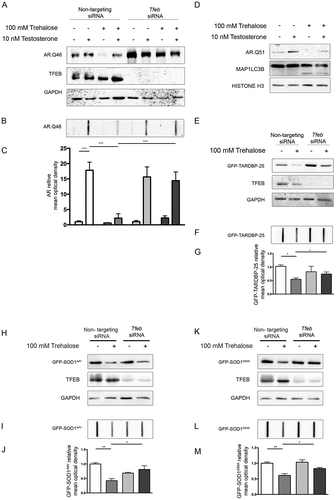
We next wanted to extend our evaluation of TFEB involvement in aggregate clearance of the protein TARDBP/TDP-43 (TAR DNA binding protein) typically characterizing sporadic forms of ALS and of fronto-lateral temporal dementia, as well as familial forms of ALS (fALS) linked to several human SOD1 (superoxide dismutase 1) mutations. In patients and cellular models of these diseases, TARDBP is cleaved releasing a neurotoxic fragment of 25 kDa (TARDBP-25/TDP-25), and we recently reported that this cleaved fragment is highly prone to aggregate into sodium dodecyl sulfate-insoluble species resistant to autophagy clearance and capable of impairing autophagy [Citation63,Citation64]. While no significant variations were found for the soluble fraction of the GFP-tagged TARDBP-25 fragment evaluated in WB (), similar to the AR model, we found that trehalose induced the clearance of the aggregated form of this fragment retained on FRA (, quantification in ). Also in this case, the pro-degradative activity of trehalose was abolished when TFEB was downregulated (), suggesting that TARDBP-25 clearance by trehalose requires the TFEB pathway. Then we studied the fALS models based on mutant SOD1, using immortalized motoneurons expressing GFP-SOD1A4V or GFP-SOD1G93A, 2 point mutations causative of fALS. We observed that trehalose induced the clearance of insoluble GFP-SOD1 mutant proteins (; quantification in ), whereas the soluble form remained similar in WB. TFEB downregulation counteracted trehalose-induced clearance of GFP-SOD1 mutant proteins. Collectively, these results indicate that the TFEB pathway is a critical mediator of autophagy clearance of misfolded proteins in NDs.
Trehalose-induced TFEB activation is mediated by PPP3
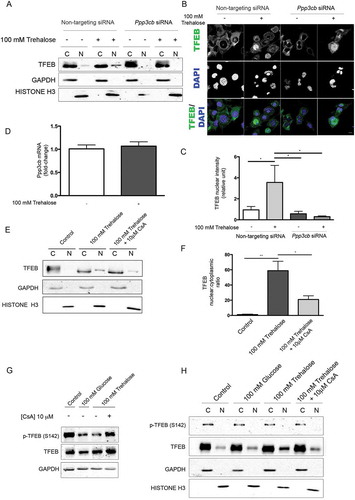
Because we demonstrated that the pro-autophagic activity of trehalose on misfolded protein requires the presence of TFEB, we searched for the molecular mechanism used by trehalose to activate TFEB. TFEB nuclear translocation and transcriptional activation is controlled through dephosphorylation by PPP3CB [Citation38–Citation40]. Thus, in trehalose-treated cells, we evaluated TFEB translocation upon downregulation of Ppp3cb. By both WB () and IF (, quantification in ), we observed that Ppp3cb downregulation strongly reduced trehalose-induced nuclear translocation of TFEB. Despite this, trehalose did not directly affect Ppp3cb levels, because no variation in mRNA levels was observed by RT-qPCR after trehalose exposure (). Interestingly, we also found that PPP3CB activity was likely to be required to mediate TFEB translocation to the nucleus in trehalose-treated cells. In fact, we observed that the immunosuppressant cyclosporin A, a potent PPP3CB inhibitor [Citation65], reduced nuclear translocation of TFEB (, quantification ), and counteracted dephosphorylation at p-Ser142 when TFEB was overexpressed (). In addition, in WB the TFEB immunoreactive band was normally upshifted in the absence of trehalose appearing as a doublet, while the ratio of the 2 TFEB bands was specifically reduced by trehalose ( and (a)), suggesting that TFEB was dephosphorylated in these conditions. These experiments indicate that PPP3CB activity is required for TFEB delocalization upon trehalose treatment.
Figure 4. Trehalose-induced TFEB activation is mediated by PPP3CB activity. (a-c) NSC34 were transfected with Ppp3cB or non-targeting siRNA, and treated with 100 mM trehalose or untreated (as control) for 48 h. (A) WB analysis of cytoplasmic (C) and nuclear extracts (N). (b) IF analysis performed with anti-TFEB antibody (green), nuclei were stained with DAPI (blue) (63X magnification). Scale bar: 10 μm. (c) The bar graph represents the quantification of TFEB nuclear intensity; the fields were randomly selected and at least 100 cells for each sample were analyzed (n = 3) (*p < 0.05, ** p < 0.005, *** p < 0.001, one-way ANOVA with Tukey’s test). (d) RT-qPCR for Ppp3cB mRNA performed on NSC34 cells treated with 100 mM trehalose or untreated (as control) for 48 h. The relative fold difference of mRNA expression was determined using untreated samples as internal control. Data are means ± SD of 4 independent samples. (e) WB analysis of cytoplasmic (C) and nuclear extracts (N) on NSC34 cells treated (or untreated) with 100 mM trehalose, in the absence or in presence of 10 μM CsA) for 1 h. (f) The bar graph represents mean ± SD for n = 4 independent samples of nuclear:cytoplasmic TFEB ratio compared to control (*p < 0.05, ** p < 0.005, one-way ANOVA with Tukey’s test). (g-h) For the determination of TFEB phosphorylation levels at Ser142, NSC34 were transfected with TFEB and treated with 100 mM glucose or 100 mM trehalose in the absence or in presence of 10 μM CsA for 1 h. (g) WB analysis and (h) WB analysis of cytoplasmic (C) and nuclear extracts (N) were performed. For WB fractionation experiments, GAPDH and histone H3 were used as an internal loading control for cytoplasmic and nuclear fraction, respectively.
Trehalose induces transient lysosome damage
PPP3CB is a Ca2+ and calmodulin-dependent Ser/Thr protein phosphatase. It has been recently shown that PPP3CB activity required for TFEB dephosphorylation can be triggered by the lysosomal fusion and other lysosomal events that lead to Ca2+ release [Citation39,Citation40]. To evaluate the effect of trehalose on lysosomes, we analyzed their morphology and abundance by electron microscopy (EM) of treated and untreated NSC34 cells. While in control cells occasional lysosomes were visible, treated cells displayed several enlarged lysosomes whose size increased after incubation with trehalose, doubling their size at 24–48 h (, quantification in ). Interestingly, at 2 and 6 h of trehalose treatment more than half of the enlarged lysosomes in treated cells also presented gaps in their membrane (, and quantification in ), suggesting that trehalose treatment compromises the integrity of the lysosome limiting membrane. Starting from 24 h, the trehalose-induced lysosomal damage became attenuated, and at 48 h the overall number of damaged lysosomes was found to be greatly reduced. Collectively, these data support the hypothesis that the trehalose induction of lysophagy triggers the clearance of damaged lysosomes and restores lysosomal homeostasis ().
Figure 5. Electron microscopy analysis of trehalose effects on lysosome morphology. (a) NSC34 cells were treated with 100 mM trehalose for the indicated times and processed for electron microscopy. Lys, lysosome. Scale bar: 500 nm. (b) Quantification of the diameter of autophagic vesicles (AV) and lysosomes (Lys) in control (CTR) and trehalose-treated cells at different times. Examples of the morphology of the quantified organelles are shown below the graph. Scale bar: 200 nm. (c) High-scale magnification of an enlarged and damaged lysosome. Arrows point to gaps in the limiting membrane. Please note that the electron-dense material in the lysosomal lumen is sparse, a feature that is never observed in untreated cells. (d) Quantification of the number of lysosomes presenting gaps on their limiting membrane in control cells and cells treated as indicated (*p < 0.05, *** p < 0.001 non-parametric one-way ANOVA with Kruskal-Wallis test). (e) WB analysis was performed on NSC34 cells treated with 100 mM trehalose or glucose (as control) for different time periods.
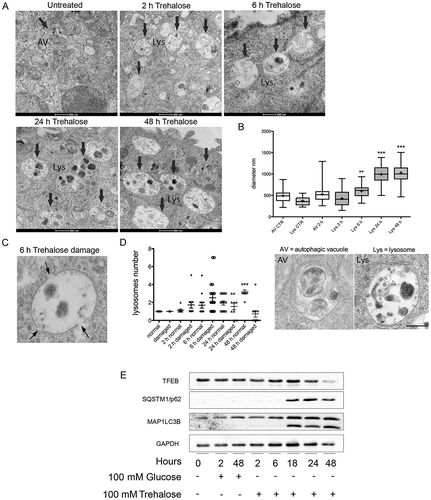
We also performed a time course analysis following the effect of trehalose on SQSTM1/p62 levels and MAP1LC3B (I vs II) activation up to 48 h. We found that, already at 18 h, trehalose strongly induced a physiological response of both autophagic markers; this activation lasted up to 48 h (). Thus, autophagy activation paralleled the TFEB nuclear translocation and activity which increased at 18 h and reached its maximal level at 48 h ().
To assess whether trehalose treatment leads to lysosome membrane permeabilization (LMP) that could cause Ca2+ release from lysosomes, we transfected NSC34 cells with a plasmid encoding EGFP-LGALS3, a fluorescent form of LGALS3/galectin 3 (lectin, galactose binding, soluble 3), which binds β-galactoside residues present in the lysosomal lumen [Citation66]. We then analyzed cells counterstained to detect the lysosomal membrane marker LAMP1 (lysosomal-associated membrane protein 1) by confocal and super-resolution microscopy. LGALS3 is normally diffuse in the cytoplasm and nucleus, but is sequestered inside lysosomes to mark individual lysosomes when LMP is induced [Citation66,Citation67]. Consistent with this, in basal conditions, we found EGFP-LGALS3 diffuse throughout the cell, while 2 and 6 h of trehalose treatment resulted in a 3-fold increase of EGFP-LGALS3-positive puncta accompanied by a global reduction of nuclear and cytoplasmic EGFP-LGALS3 in its diffuse form. As a positive control to induce LMP, we treated with the compound L-leucyl-L-leucine methyl ester (LLOMe) [Citation68], which resulted in significant puncta formation (). From 18 h to 48 h of trehalose treatment, EGFP-LGALS3 puncta per cells decreased until levels similar to basal conditions (). WB analysis confirmed that after an initial accumulation of EGFP-LGALS3 (2 and 6 h), the protein levels decreased during the time period of treatment, indicating the clearance of the protein (). By STED microscopy, we observed the appearance of ring-like EGFP-LGALS3 structures, positive for LAMP1 in cells treated for 6 h with trehalose (), indicating that lysosomes are permeabilized.
Figure 6. Trehalose induces lysosome membrane permeability. NSC34 cells were transfected with a plasmid encoding EGFP-LGALS3, and treated with 100 mM trehalose for different time periods or with 0.3 mM LLOMe for 1 h as a positive control. (a) Fluorescence microscopy analysis on cells treated with trehalose for 2 or 6 h, or with LLOMe (63X magnification). Scale bar: 10 μm. (b) The bar graph shows the quantification of percentage of cells with > 3 EGFP-LGALS3 puncta after trehalose treatment at different time points; the fields were randomly selected and at least 100 cells for each sample were counted (n = 3). (** p < 0.005, *** p < 0.001, one-way ANOVA with Tukey’s test.) (c) WB analysis of EGFP-LGALS3 protein levels, GAPDH was used as a loading control (d) Cells treated as in A and labeled with anti-LAMP1 (red) to visualize lysosomes were analyzed by STED microscopy. Arrows point to examples of EGFP-LGALS3 (green)-positive lysosomes. Scale bar: 10 μm (e) Cytofluorimetric analysis performed on NSC34 cells treated with trehalose for different time periods, and labelled with LysoTracker Green. Mean fluorescence intensity was measured (n = 4) (*** p < 0.001, one-way ANOVA with Tukey’s test).
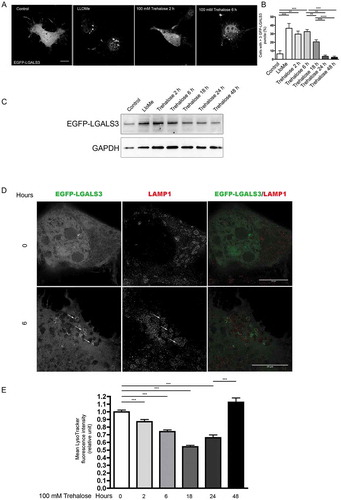
Strikingly, we observed a gradual decrease between 2 and 18 h of trehalose treatment in fluorescence associated with the dye LysoTracker Green that binds H+ in the lysosomal lumen, which is reverted by 48 h of treatment (). Overall, these data indicate that trehalose induces lysosomal enlargement and damage, leading to an increase in LMP in treated cells.
The analogs melibiose and lactulose mimic the pro-autophagic effects of trehalose
Trehalose effects have already been assayed in several different mouse models of NDs linked to misfolded protein neurotoxicity with promising results [Citation11,Citation15–Citation19,Citation21–Citation25]. However, a large fraction of trehalose is degraded by intestinal TREH (trehalase [brush-border membrane glycoprotein]), when this compound is taken orally [Citation69]. Thus, if approved for human use, trehalose is likely to be inefficient, restricting its use to intravenous injections. To circumvent such potential limitation, we evaluated whether trehalose analogs, already described as TREH-resistant and capable of activating autophagy and to remove misfolded protein species [Citation70,Citation71], trigger responses similar to those observed here upon trehalose treatment. Interestingly, we found that both melibiose and lactulose stimulated nuclear translocation of endogenous TFEB, both in IF () and in cell fractionation assay in WB (, quantification in ). In addition, induction of TFEB translocation obtained both with melibiose and lactulose was partially reverted by cyclosporin A (, respectively).
Figure 7. Melibiose and lactulose induce TFEB nuclear translocation. (a) IF analysis of TFEB localization performed on NSC34 cells treated with 100 mM trehalose, 100 mM melibiose or 100 mM lactulose for 48 h. Nuclei were stained with DAPI (blue) (63X magnification). Scale bar: 10 μm. (b) WB analysis of cytoplasmic (C) and nuclear extracts (N) on NSC34 cells untreated (control) or treated with 100 mM melibiose or 100 mM lactulose for 48 h. GAPDH and histone H3 were used as internal loading control for cytoplasmic and nuclear fractions, respectively. (c) The bar graph represents the mean ± SD for n = 4 independent samples of nuclear:cytoplasmic TFEB ratio compared to untreated cells (*p < 0.05 one-way ANOVA with Tukey’s test).
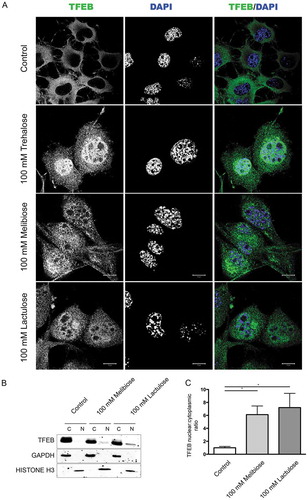
Figure 8. Melibiose and lactulose effects are mediated by PPP3CB: induction of ALP gene expression and LMP. (a-b) WB analysis of cytoplasmic (c) and nuclear extracts (N) of NSC34 cells untreated (control) or treated with 100 mM melibiose (a) or 100 mM lactulose (b) in the absence or presence of 10 μM CsA for 1 h. GAPDH and histone H3 were used as internal loading controls for cytoplasmic and nuclear fractions, respectively. (c-h) RT-qPCR on NSC34 cells untreated (control) or treated with 100 mM trehalose, 100 mM melibiose or 100 mM lactulose for 48 h. The relative fold difference of mRNA expression was determined using untreated samples as internal control. Data are means ± SD of 4 independent samples. RT-qPCR for the following mRNA: Tfeb (c); Zkscan3 (d); Sqstm1/p62 (e); Map1Lc3b (f); Hspb8 (g); Bag3 (h). Bar graphs represent the relative fold induction of these genes (*p < 0.05, ** p < 0.005, *** p < 0.001, one-way ANOVA with Tukey’s test). (i) Fluorescence microscopy analysis (63X magnification) performed on NSC34 cells that were transfected with a plasmid encoding EGFP-LGALS3, and treated with 100 mM trehalose, 100 mM melibiose or 100 mM lactulose for 2 h; scale bar: 10 μm. (j) Bar graph shows the quantification of percentage of cells with > 3 EGFP-LGALS3 puncta; the fields were randomly selected and at least 100 cells for each sample were counted (n = 3) (** p < 0.005, *** p < 0.001, one-way ANOVA with Tukey’s test).
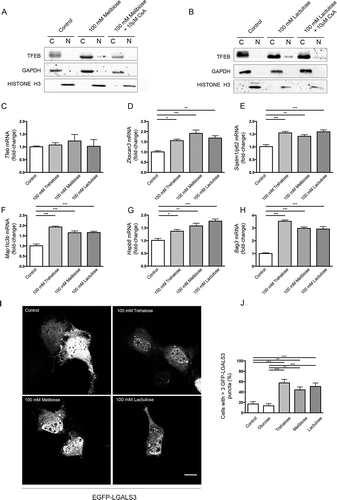
This finding suggests that, as is the case with trehalose, upon melibiose or lactulose treatment, the phosphatase activity of PPP3CB might be required for induction of TFEB activation, but Tfeb de novo transcription was not required (). Both TREH-resistant molecules induced the expression of Zkscan3 (), Sqstm1/p62 (), Map1Lc3b (), Hspb8 () and Bag3 () with an intensity analogous to that observed in trehalose treatments at the same doses. Finally, both compounds induced EGFP-LGALS3-positive puncta to a similar extent to trehalose (), indicating that they are also able to induce LMP.
To assess the pro-degradative activity of melibiose and lactulose in comparison to trehalose, we performed FRA analysis. We observed that both trehalose analogs were able to revert AR.Q46 aggregation, with effects comparable to trehalose (, quantification in ). Melibiose and lactulose were able to induce the clearance of soluble ARpolyQ, effects fully counteracted by TFEB downregulation (, respectively), suggesting that their pro-degradative activities require this transcription factor as we showed in the case of trehalose.
Figure 9. TFEB mediated pro-degradative effects of melibiose and lactulose on ARpolyQ clearance. (a) FRA analysis performe on NSC34 cells transfected with AR.Q46, untreated or treated with 100 mM trehalose, melibiose or lactulose in the absence or in presence of 10 nM testosterone for 48 h. (b) The bar graph represents the mean relative optical density of FRA ± SD for n = 3 independent samples (*** p < 0.001, two-way ANOVA with Bonferroni’s test). (c-d) WB analysis performed on NSC34 cells transfected with Tfeb siRNA or non-targeting siRNA, and with AR.Q46, in absence or in presence of 10 nM testosterone for 48 h treated with 100 mM melibiose or lactulose, respectively.
In summary, all the experiments performed using the TREH-resistant analogs of trehalose proved that they act via LMP and TFEB, suggesting that they might be effective alternatives to trehalose.
Discussion
Trehalose is a natural disaccharide used in food production that recently was found able to enhance the autophagic removal of neurotoxic misfolded proteins prone to aggregate [Citation7–Citation14], ameliorating disease phenotype in several cell and mouse models of NDs (including human tauopathy, HD, PD, ALS, oculopharyngeal muscular dystrophy) [Citation13,Citation15–Citation26]. Trehalose stimulates the nuclear translocation of TFEB and the expression of genes regulating autophagy [Citation9–Citation11,Citation13,Citation27–Citation30]. To trigger autophagy, trehalose must be imported inside the cells by SLC2A8/GLUT8 [Citation31,Citation32], and its action does not involve the MTOR pathway [Citation9,Citation16,Citation24,Citation28,Citation30]. We excluded any direct activity of trehalose on TFEB expression, while trehalose may indirectly control the TFEB phosphorylation state and nuclear translocation, via the adenosine 5ʹ-monophosphate-activated protein kinase pathway [Citation32] or the AKT pathway [Citation30]. However, it is unclear how p-TFEB is dephosphorylated. So far, the only phosphatase reported to be directly able to dephosphorylate TFEB, allowing its activation, is PPP3CB [Citation39,Citation40]. Here, we showed that trehalose has no effects on Ppp3cb expression, while genetic or pharmacological blockage of PPP3CB strongly reduces trehalose-induced dephosphorylation at p-Ser142 of TFEB and TFEB nuclear translocation. Therefore, trehalose acts on TFEB by modulating PPP3CB enzymatic phosphatase activity, which is required to allow trehalose-mediated activation of TFEB. We propose a mechanism by which this might occur.
Trehalose induces rapid, partial and transient lysosomal damage and permeabilization, with Ca2+ release and PPP3CB-induced TFEB nuclear translocation. Subsequent induction of autophagy, perhaps to heal lysosomal damage, might lead to activation of autophagy and clearance of aggregated neurotoxic deposits, possibly as a side effect ().
Figure 10. Proposed model of the mechanism of action trehalose, and its analogs: trehalose, melibiose and lactulose act on lysosomes inducing a transient lysosomal membrane permeability. This causes Ca2+ leaks which activates the phosphatase PPP3/calcineurin. PPP3 specifically de-phosphorylates TFEB inducing its activation and nuclear translocation to activate autophagy and lysophagy with removal of damaged lysosomes and in parallel (if present) misfolded protein aggregates.
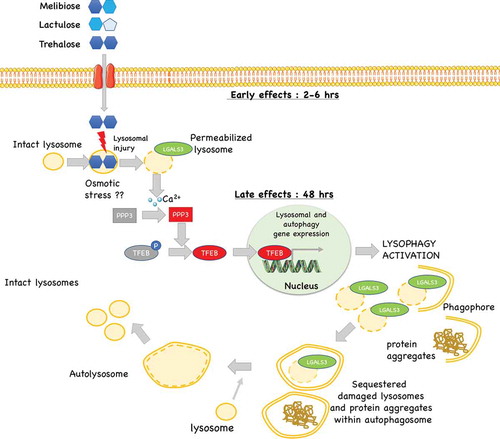
To support our model we found, to our surprise, that trehalose induces transient changes in lysosomal morphology, characterized by enlargement and formation of gaps in lysosome membranes in as much as half of the cell lysosomes. We demonstrate that such changes were associated with a LMP characterized by incorporation of LGALS3 and transient loss of LysoTracker fluorescence. Notably, we observed that all these events occur earlier than TFEB activation, suggesting that they might indeed be the cause of TFEB activation. Interestingly, the reduction of LysoTracker-positive lysosomes has already been linked to increased LMP and release of Ca2+ ions [Citation38–Citation40]. In line, in mouse models of retinitis pigmentosa, trehalose triggers LMP and autophagy [Citation72]. Damaged lysosomes are specifically sequestered and become the target of a lysosome-specific form of autophagy, called lysophagy [Citation67,Citation68,Citation73–Citation76]. Pharmacological induction of LMP has been previously shown to trigger oxidative stress, and inflammation, ultimately leading to apoptosis or necrosis [Citation68,Citation75]. However, it has been reported that a transient damage on a limited pool of lysosomes can be reversible and may specifically induce rapid lysophagy [Citation66]. Thus, lysophagy is fundamental to sequester damaged lysosomes restoring the normal pool of lysosomes via a TFEB-dependent lysosomal biogenesis.
How could trehalose cause LMP? Our ultrastructural analysis indicates that, in trehalose-treated cells, a fraction of lysosomes are bloated and present evident gaps in their limiting membrane. Thus, trehalose might trigger LMP by lysosomal osmotic stress which will release lysosomal Ca2+ activating PPP3 and consequently TFEB dephosphorylation and activation. Whether trehalose is imported into lysosomes to cause osmotic stress remains to be determined. While this mechanism removes damaged lysosomes via lysophagy, the associated upregulation of autophagy regulators is used to clear other damaged or aberrant components from neurons, most notably polyQ-containing proteins, or other ND-associated misfolded proteins. Alteration of lysosomal dynamics have also been described in PD. Indeed, lysosomal breakdown and accumulation of autophagosomes is detectable in PD brain samples in association with Lewy bodies. TFEB activation restores the number of lysosomes via an increased turnover of autophagosomes [Citation34]. Thus, trehalose might exert a protective function in PD primarily because of its ability to activate TFEB restoring normal lysosomal function.
We observed that trehalose induces, in a TFEB-dependent manner, the expression of the genes coding for BECN1, ATG10, ATG12, SQSTM1/p62 and MAP1LC3B, which are all required for autophagy activation, as well as of PPARGC1A, which is both a target and an inducer of TFEB expression [Citation77]. Trehalose also induced Zkscan3 expression, which acts as a physiological TFEB antagonist. This effect was independent of TFEB activation and it might be a feedback mechanism to restore basal autophagy levels with pathways different from those controlled by TFEB. Trehalose also modulated key components (HSPB8 and BAG3) of the CASA complex which delivers misfolded proteins to autophagosomes. Here, TFEB was marginally involved, but HSPB8, SQSTM1/p62, MAP1LC3B and ATG12 levels all increase 48 h after trehalose treatment, indicating that other pathways might coordinate the response to the drug. It is the combined activities of these regulators that ultimately enhance autophagic clearance of the insoluble ARpolyQ, TARDBP and mutant SOD1 species causative of neurodegeneration [Citation51].
Unfortunately, in human, trehalose cannot be given orally because it is degraded by the TREH present in the gastrointestinal tract. Notably, the lack of TREH enzymatic activity in the gut, detected in the Greenland population, has no consequence on blood glucose concentration and has no impact on human health [Citation69]. This suggests that trehalose could be potentially administered intravenously, but this would require its registration as an approved drug and it would likely decrease compliance of patients to a treatment that can be currently provided as a supplement of a natural compound. We showed that the analogs of trehalose, melibiose and lactulose, which are resistant to intestinal TREH, recapitulated all its effects, indicating that they are valid and potent mimics of trehalose and could be highly beneficial to the treatment of NDs.
In summary, lysosomal damage underlies TFEB-mediated activation of aggregate clearance induced by trehalose and such a mechanism is likely crucial to trigger neuroprotection in a number of currently incurable human diseases.
Material and methods
Chemicals
The following compounds were used: D-(+)-trehalose dihydrate (100 mM, T9531), melibiose (100 mM, M5500), lactulose (100 mM, L7877), D-(+)-glucose (100 mM, 49159), cyclosporin A (10 µM, 30024), testosterone (10 nM, 86500), Leu-Leu methyl ester hydrobromide (LLOMe; 0.3 mM, L7393), CHIR 98024 (3 μM, SML1094), ascorbic acid (AA; 200 μM, A5960), retinoic acid (1 μM, K2625), and dbcAMP (100 μM, D0627) were purchased from Sigma-Aldrich. SB431542 (10 µM, 1614/1), SAG (0.5 μM, 4366/10), and PMA (0.5 μM, 10009634) were purchased from Tocris Bioscience. LDN 193189 (0.2 mΜ, S2618) was purchased from Selleckchem. BDNF (2 ng/mL, 450–02), GDNF (5 ng/mL, 450–10), TGFB1 (1 ng/mL, 100–21) and activin A (2 ng/mL, 120–14) were purchased from Protein Tech.
Plasmids
TFEB-pCDNA3 plasmid was kindly provided by Prof. A. Ballabio (Telethon Institute of Genetics and Medicine (TIGEM), Dulbecco Telethon Institute, Federico II University, Naples) [Citation40]. EGFP-LGALS3 plasmid was kindly provided by Prof. M. A. Jäättelä (Danish Cancer Society Research Center, Copenhagen, Denmark) [Citation66]. The AR.Q46 plasmid codes for the mutant human AR with 46 CAG repeat has been originally provided by Prof. M. Marcelli (Baylor College of Medicine, Houston, TX, USA) and already described in [Citation55]. GFP-TARDBP-25/TDP-25 codes for the TARDBP C-terminal 25-kDa fragment fused with the GFP protein. The plasmid was kindly provided by Prof. L. Petrucelli (Mayo Clinic, Jacksonville, FL, USA) [Citation78]. GFP-SOD1A4V and GFP-SOD1G93A were obtained by cloning the SOD1 gene with point mutations in the pEGFP plasmid (Clontech, 6084-1) [Citation62,Citation79].
Cell cultures and transfections
The mouse motoneuron-like hybrid cell line (NSC34) [Citation80,Citation81] was cultured in DMEM high-glucose medium (EuroClone, ECB7501L) supplemented with 5% fetal bovine serum (Sigma-Aldrich, F7524), 1 mM L-glutamine (EuroClone, ECB3004D), and antibiotics (penicillin, SERVA, 31749.04; streptomycin, SERVA, 35500.01), and grown at 37°C in 5% CO2. Cells treated with sugars were seeded in DMEM low glucose (EuroClone, ECM0749L) to avoid hyperosmotic shock.
NSC34 were transfected with 0.7 μg of DNA plasmid using Lipofectamine® (ThermoFisher Scientific, 18324012) and TRF/transferrin (Sigma-Aldrich, T8150).
In all the silencing experiments, NSC34 cells were transfected with 20–40 pmole Tfeb siRNA (sense: 5ʹ GGAUCAAGGAGCUGGGAAUUU 3ʹ, antisense: 5ʹ AUUCCCAGCUCCUUGAUCCUU 3ʹ), Ppp3cb siRNA (sense: 5ʹ UGACAGAAAUGUUGGUAAAUU 3ʹ; antisense: 5ʹ UUUACCAACAUUUCUGUCAUU 3ʹ) or non-targeting siRNA (sense: 5ʹ UAGCGACUAAACACAUCAAUU 3ʹ; antisense: 5ʹ UUGAUGUGUUUAGUCGCUAUU 3ʹ) using Lipofectamine® 2000 (ThermoFisher Scientific, 11668019). All siRNAs were purchased from Dharmacon.
IPSCs cultures and differentiation
iPSCs were obtained in the lab of Dr. Kennet Fischbeck and Dr. Christopher Grunseich (NIH, Bethesda, MD, USA) from SBMA patient fibroblasts. iPSCs were cultured on Matrigel (Corning, 354277) and fed with E8 (ThermoFisher Scientific, A1517001). Every 5–6 days, cells were split using Accutase (ThermoFisher Scientific, 00-4555-56), and in the first day, E8 was supplemented with ROCK-I (5 μM; Selleckchem, S1049).
Generation of small molecules neural progenitor cells and differentiation towards motor neurons
iPSCs were differentiated into small molecules neural progenitor cells (smNPCs) following the already published protocol [Citation82]. In brief, embryo bodies were derived from iPSCs and were maintained in a medium composed of DMEM-HAM’S F-12 (Euroclone, ECM0090L), Neurobasal medium (ThermoFisher Scientific, 21103049), N-2™ supplement (ThermoFisher Scientific, 17502048) and B-27™ supplement (ThermoFisher Scientific, 17504044) and antibiotics (N2B27 medium). Cells were treated with CHIR, SB431542, LDN 193189, SAG, and AA. After 6 days, embryo bodies were dissociated and plated on Matrigel (hESC-qualified Matrix; Corning®, 354277) in a medium composed of N2B27 with CHIR, AA, and PMA. smNPCs were cultured for 7–8 passages before starting differentiation into motoneurons. Motoneuron differentiation was achieved by plating smNPCs on Matrigel-coated plates at 300,000 cell/mL in N2B27 medium supplemented with AA, CHIR, BDNF, GDNF, and retinoic acid. After 6 days, Patterning medium was changed to Maturation medium (N2B27 supplemented with dbCAMP, AA, GDNF, BDNF, TGFB1, and Activin A (only in the first two days). After two days, the cells were split and plated at 200,000 cell/well in a 12-well plate, and maintained in maturation medium up to 10 days.
Real-time quantitative polymerase chain reaction (RT-qPCR)
For RT-qPCR, cells were seeded at 180,000 cell/well in 6-well plates transfected/treated as explained in the text, and incubated for 48 h. Total RNA was extracted using Tri-Reagent (Sigma-Aldrich, T9424) following the manufacturer’s protocol. RNA was quantified using a NanoDrop 2000 (ThermoFisher Scientific, ND-2000) and 1 μg per sample was treated with DNAse and reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (ThermoFisher Scientific, 4368814). The primer sequences are listed in Table S1.
RT-qPCR was carried out using the CFX96 Real Time System (Bio-Rad Laboratories), the iTaq SYBR Green Supermix (Bio-Rad Laboratories, 1725124), and with 500 nmol primers. Data were normalized using Rplp0. The experiments were performed with 4 independent samples (n = 4).
Western blotting and filter retardation assay
For WB and FRA, cells were seeded at 90,000 cell/well in 12-well plates, transfected/treated as explained in the text and incubated for 48 h. The cells were collected and centrifuged at 100 g for 5 min. at 4°C. The pellets were resuspended in phosphate-buffered saline (PBS; Sigma-Aldrich, P4417) supplemented with Protease Inhibitor Cocktail (Sigma-Aldrich, P8340) and an ultrasonic homogenization was performed. The protein concentration was quantified with the bicinchoninic acid method using a Quantum Protein Assay Kit (Euroclone, EMP014500).
For WB analysis, 15 μg of total proteins was loaded, and 10% (or 15%) sodium dodecyl sulfate-polyacrylamide gel electrophoresis was performed. The gels were electro-transferred to a nitrocellulose membrane using a Trans-turbo transfer System (Bio-Rad Laboratories, 1704150).
For FRA, 1.5 μg (for AR.Q46), 9 μg (for GFP-TARDBP-25) or 6 μg (for GFP-SOD1 plasmids) of total proteins were filtered on a 0.2-μm cellulose acetate membrane (Whatman GE Healthcare, GEH10404180) using a Bio-Dot SF Microfiltration Apparatus (Bio-Rad Laboratories, 1703938).
To perform nuclear-cytoplasmic TFEB localization by WB, NSC34 were seeded at 180,000 cell/well in 6-well plates, transfected/treated as explained in the text and incubated for 48 h. The cells were collected and centrifuged at 375 g for 5 min. at 4°C. The pellets were lysed in Triton X-100 lysis buffer (50 mM Tris-HCl, pH 7.5, 0.5% Triton X-100 (Sigma-Aldrich, X100), 137.5 mM NaCl, 10% glycerol (Sigma-Aldrich, G5516), 5 mM ethylenediaminetetraacetic acid) containing protease inhibitor cocktail (Sigma-Aldrich, P8340) and the nuclear-cytoplasmic fractions were isolated as described previously [Citation77].
The membranes obtained by WB or FRA were immunoblotted using the following antibodies: anti-SQSTM1/p62 (Abcam, ab91526; 1:2,000); anti-LC3A/B (Sigma-Aldrich, L8918, 1:2,000); anti-TFEB (Bethyl Laboratories, A303-673A, 1:4,000), anti-phospho-TFEB (Ser142; Merck-Millipore, ABE1971; 1:3,000), anti-GAPDH (FL-335; Santa-Cruz Biotechnology, sc-25778; 1:3,000), anti-AR (H280; Santa Cruz Biotechnology, sc-13062; 1:1,000), anti-histone H3 (Abcam, ab1791; 1:40,000), anti-GFP (Abcam, AB1218; 1:2,000). The primary antibodies were prepared in 5% non-fat dried milk (Euroclone, EMR180500) in TBS-T (Tris base 20 mM, NaCl 140 mM, pH 7.6 and 0.01% Tween 20 [Sigma-Aldrich, P1379]).
Immunoreactivity was detected using secondary peroxidase conjugated goat anti-rabbit IgG-HRP (Jackson Immunoresearch Laboratories, 111–035-003: 1:5,000) and enhanced chemiluminescent (ECL) detection reagent (Clarity™ ECL western Blotting substrate; Bio-Rad Laboratories, 1705060). Images were acquired using a Chemidoc XRS System (Bio-Rad Laboratories, 1708265) and densitometric quantification was performed using Image Lab Software, version 5.2.1 (Bio-Rad Laboratories).
Immunofluorescence analysis (IF)
For IF analysis, the cells were seeded at 35,000 cells/well in 24-well plates on 13-mm coverslips, transfected/treated as explained in the text and incubated for 48 h. The cells were fixed with 4% paraformaldehyde in PBS and then permeabilized in 0.2% Triton X-100 in PBS for 10 min. The cells were incubated with primary antibody anti-TFEB (Bethyl Laboratories, 1:400) in 5% non-fat dried milk- TBS-T blocking solution, and then with secondary antibody goat anti-rabbit Alexa Fluor® 488 (ThermoFisher Scientific, A11070; 1:1,000) in 5% non-fat dried milk (EuroClone, EMR0145000) TBS-T blocking solution. Nuclei were stained with DAPI (1:10,000 in PBS). Coverslips were mounted onto slides with Mowiol® 4-88 (Merck-Millipore, 475904) and analyzed with an LSM510 Meta system confocal microscope (Zeiss) and images were processed with the Aim 4.2 software (Zeiss).
Image analysis
TFEB nuclear intensity was measured using ImageJ software. For each treatment, 100 cells were analyzed and a value of intensity per nucleus was measured.
Measurement of lysosomal membrane permeabilization by lysosomal LGALS/galectin puncta assay
To perform a lysosomal LGALS/galectin puncta assay, NSC34 were plated at 35,000 cell/well in 24-well plates on 13-mm coverslips and transfected with 0.4 μg of GFP-LGALS3/Galectin3 plasmid. The cells were treated with 100 mM trehalose, melibiose, lactulose or glucose (as negative control) for different time periods. LLOMe was included as a positive control for LMP (0.3 mM for 1 h). The cells were fixed as described above, and quantification of cells with > 3 EGFP-LGALS3 puncta was performed by manual counting of 3 fields per sample, using a PL 10X/20 eyepiece with graticules (100 mm × 10 mm in 100-grid divisions) as described in [Citation66]. The fields were randomly selected and at least 100 cells for sample were counted (n = 3).
Measurement of lysosomal membrane permeabilization by cytometer analysis
NSC34 cells were seeded at 90,000 cell/well in 12-well plates and treated with 100 mM trehalose for different time periods (from 2 to 48 h). The cells were incubated with 100 nM of the lysosomotropic probe LysoTracker Green DND-26 (ThermoFisher Scientific, L7526) for 30 min. The cells were collected, resuspended in 4% FBS in PBS and analyzed on a NovoCyte flow cytometer (Acea Biosciences, Inc.). Mean LysoTracker fluorescence intensity was recorded from 50,000 cells for each sample (n = 4).
Stimulated emission depletion microscopy (STED)
NSC34 cells were plated at 35,000 cell/well in 24-well plates on 13-mm coverslips and transfected with 0.4 μg of GFP-LGALS3/Galectin3 plasmid. The cells were treated with 100 mM trehalose for 6 h. The cells were fixed for 25 min with 4% paraformaldehyde in PBS at 37°C and then permeabilized in 0.2% Triton X-100 in PBS for 10 min. The cells were incubated with primary antibody anti-LAMP1 (Abcam, AB24170; 1:1,000) in 5% non-fat dried milk-TBS-T blocking solution, and then with secondary antibody anti-rabbit IgG-Atto647N (Sigma-Aldrich, 40839; 1:400) in 5% non-fat dried milk-TBS-T blocking solution. Coverslips were mounted onto slides with Mowiol® 4-88 (Merck-Millipore, 475904) and analyzed with a Leica TCS SP8 STED 3X with 3 depletion lines (592, 660 and 775 nm), with HC PL APO 100X/1.40 oil objective. Images were acquired through the Software Leica LAS X and processed using ImageJ (version 1.51).
Electron microscopy analysis
NSC34 cells were seeded at 180,000 cell/well in a 2-well Nunc® Lab-Tek®Chamber Slide™ system (Nunc, C6682). The cells were treated with 100 mM trehalose for different times, and then fixed using 2.5% glutaraldehyde (Sigma-Aldrich, G7776) in 0.1 M sodium cacodylate buffer (Sigma-Aldrich, C0250), pH 7.4 for 1 h at room temperature. The cells were postfixed in osmium tetroxide (Electron Miscroscopy Science, 19100) for 2 h, and 1% uranyl acetate (SERVA Electrophoresis, 77870) for 1 h. Subsequently, samples were dehydrated through a graded ethanol series and flat embedded in resin EMBED-812 (Electron Microscopy Science, 14120) for 24 h at 60°C. Ultrathin sections (50 nm) were cut parallel to the substrate, stained with 5% uranyl acetate in 50% ethanol and observed with a CM10 electron microscope (Philips, Eindhoven, The Netherlands). Digital images were taken with a Megaview 3 camera. Analysis of morphologically identified double-membrane autophagic vacuoles (AV) and single-membrane lysosomes (Lys) number and diameters was assessed in 10 cells for each treatment. The diameter of these structures was measured with an iTEM software package (Olympus-SYS; Olympus Corporation) and plotted as box plots [Citation83].
Statistical analysis
The data are presented as mean ± SD. Statistical analysis has been performed by using Student’s t-test to compare 2 groups. To compare 3 or more groups were made analysis of variance (ANOVA): One-Way ANOVA to compare the effect of one variable or Two-Way ANOVA to compare the effect of 2 independent variables. P value <0.05 was considered statistically significant. When ANOVA was significant, we performed a post hoc test (see figure legends for details). Analyses were done with the PRISM (version 5) software (GraphPad Software). Statistical analysis for the EM experiments was performed using the Mann-Whitney test and Kruskal-Wallis test, means were considered significantly different if P < 0.05.
Supplemental Material
Download MS Excel (11.3 KB)Acknowledgments
We are grateful to Prof. Maria Jäättelä (Danish Cancer Society Research Center, Copenhagen, Denmark) for the GFP-LGALS3/Galectin3 plasmid, to Prof. Leonard Petrucelli (Mayo Clinic, Jacksonville, FL, USA) for the GFP-TARDBP-25 plasmid, to Prof. Andrea Ballabio (Telethon Institute of Genetics and Medicine (TIGEM), Dulbecco Telethon Institute, Federico II University, Naples) for the TFEB plasmid, to Prof. Marco Marcelli (Baylor College of Medicine, Houston, TX, USA) for the AR.Q46 plasmid.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222.
- Galluzzi L, Bravo-San Pedro JM, Levine B, et al. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discov. 2017;16:487–511.
- Jackson MP, Hewitt EW. Cellular proteostasis: degradation of misfolded proteins by lysosomes. Essays Biochem. 2016;60:173–180.
- Haidar M, Timmerman V. Autophagy as an emerging common pathomechanism in inherited peripheral neuropathies. Front Mol Neurosci. 2017;10:143.
- Lumkwana D, Du Toit A, Kinnear C, et al. Autophagic flux control in neurodegeneration: progress and precision targeting-Where do we stand? Progr Neurobiol. 2017;153:64–85.
- Menzies FM, Fleming A, Caricasole A, et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron. 2017;93:1015–1034.
- Sarkar S, Rubinsztein DC. Huntington’s disease: degradation of mutant huntingtin by autophagy. FEBS J. 2008;275:4263–4270.
- Sarkar S, Rubinsztein DC. Small molecule enhancers of autophagy for neurodegenerative diseases. Mol Biosyst. 2008;4:895–901.
- Sarkar S, Davies JE, Huang Z, et al. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem. 2007;282:5641–5652.
- Rusmini P, Crippa V, Giorgetti E, et al. Clearance of the mutant androgen receptor in motoneuronal models of spinal and bulbar muscular atrophy. Neurobiol Aging. 2013;34:2585–2603.
- Lan DM, Liu FT, Zhao J, et al. Effect of trehalose on PC12 cells overexpressing wild-type or A53T mutant alpha-synuclein. Neurochem Res. 2012;37:2025–2032.
- Wang X, Fan H, Ying Z, et al. Degradation of TDP-43 and its pathogenic form by autophagy and the ubiquitin-proteasome system. Neurosci Lett. 2010;469:112–116.
- Aguib Y, Heiseke A, Gilch S, et al. Autophagy induction by trehalose counteracts cellular prion infection. Autophagy. 2009;5:361–369.
- Casarejos MJ, Perucho J, Lopez-Sendon JL, et al. Trehalose improves human fibroblast deficits in a new CHIP-mutation related ataxia. PloS one. 2014;9:e106931.
- He Q, Koprich JB, Wang Y, et al. Treatment with trehalose prevents behavioral and neurochemical deficits produced in an AAV alpha-synuclein rat model of Parkinson’s disease. Mol Neurobiol. 2016;53:2258–2268.
- Zhang X, Chen S, Song L, et al. MTOR-independent, autophagic enhancer trehalose prolongs motor neuron survival and ameliorates the autophagic flux defect in a mouse model of amyotrophic lateral sclerosis. Autophagy. 2014;10:588–602.
- Sarkar S, Chigurupati S, Raymick J, et al. Neuroprotective effect of the chemical chaperone, trehalose in a chronic MPTP-induced Parkinson’s disease mouse model. Neurotoxicology. 2014;44:250–262.
- Du J, Liang Y, Xu F, et al. Trehalose rescues Alzheimer’s disease phenotypes in APP/PS1 transgenic mice. J Pharm Pharmacol. 2013;65:1753–1756.
- Castillo K, Nassif M, Valenzuela V, et al. Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy. 2013;9:1308–1320.
- Schaeffer V, Lavenir I, Ozcelik S, et al. Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain. 2012;135:2169–2177.
- Schaeffer V, Goedert M. Stimulation of autophagy is neuroprotective in a mouse model of human tauopathy. Autophagy. 2012;8:1686–1687.
- Perucho J, Casarejos MJ, Gomez A, et al. Trehalose protects from aggravation of amyloid pathology induced by isoflurane anesthesia in APP(swe) mutant mice. Curr Alzheimer Res. 2012;9:334–343.
- Rodriguez-Navarro JA, Rodriguez L, Casarejos MJ, et al. Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiol Dis. 2010;39:423–438.
- Davies JE, Sarkar S, Rubinsztein DC. Trehalose reduces aggregate formation and delays pathology in a transgenic mouse model of oculopharyngeal muscular dystrophy. Hum Mol Genet. 2006;15:23–31.
- Tanaka M, Machida Y, Niu S, et al. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med. 2004;10:148–154.
- Debnath K, Pradhan N, Singh BK, et al. Poly(trehalose) nanoparticle prevents amyloid aggregation and suppress polyglutamine aggregation in huntington’s disease model mouse. ACS Appl Mater Inter. 2017;9:24126–24139.
- Giorgetti E, Rusmini P, Crippa V, et al. Synergic prodegradative activity of Bicalutamide and trehalose on the mutant androgen receptor responsible for spinal and bulbar muscular atrophy. Hum Mol Genet. 2015;24:64–75.
- Chen X, Li M, Li L, et al. Trehalose, sucrose and raffinose are novel activators of autophagy in human keratinocytes through an mTOR-independent pathway. Sci Rep. 2016;6:28423.
- Montie HL, Merry DE. Autophagy and access: understanding the role of androgen receptor subcellular localization in SBMA. Autophagy. 2009;5:1194–1197.
- Palmieri M, Pal R, Nelvagal HR, et al. mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat Commun. 2017;8:14338.
- Mardones P, Rubinsztein DC, Hetz C. Mystery solved: trehalose kickstarts autophagy by blocking glucose transport. Sci Signal. 2016;9:fs2.
- DeBosch BJ, Heitmeier MR, Mayer AL, et al. Trehalose inhibits solute carrier 2A (SLC2A) proteins to induce autophagy and prevent hepatic steatosis. Sci Signal. 2016;9:ra21.
- Mayer AL, Higgins CB, Heitmeier MR, et al. SLC2A8 (GLUT8) is a mammalian trehalose transporter required for trehalose-induced autophagy. Sci Rep. 2016;6:38586.
- Dehay B, Bove J, Rodriguez-Muela N, et al. Pathogenic lysosomal depletion in Parkinson’s disease. J Neurosci. 2010;30:12535–12544.
- Sardiello M, Palmieri M, di Ronza A, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–477.
- Medina DL, Fraldi A, Bouche V, et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell. 2011;21:421–430.
- Palmieri M, Impey S, Kang H, et al. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet. 2011;20:3852–3866.
- Settembre C, Ballabio A. TFEB regulates autophagy: an integrated coordination of cellular degradation and recycling processes. Autophagy. 2011;7:1379–1381.
- Medina DL, Ballabio A. Lysosomal calcium regulates autophagy. Autophagy. 2015;11:970–971.
- Medina DL, Di Paola S, Peluso I, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015;17:288–299.
- Valero T. Mitochondrial biogenesis: pharmacological approaches. Cur Pharm Design. 2014;20:5507–5509.
- Sanchis-Gomar F, Garcia-Gimenez JL, Gomez-Cabrera MC, et al. Mitochondrial biogenesis in health and disease. Molecular and Therapeutic Approaches. Cur Pharm Design. 2014;20:5619–5633.
- Dorn GW II, Vega RB, Kelly DP. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Gene Dev. 2015;29:1981–1991.
- Chauhan S, Goodwin JG, Chauhan S, et al. ZKSCAN3 is a master transcriptional repressor of autophagy. Molecular Cell. 2013;50:16–28.
- Arndt V, Dick N, Tawo R, et al. Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr Biol. 2010;20:143–148.
- Kettern N, Rogon C, Limmer A, et al. The Hsc/Hsp70 co-chaperone network controls antigen aggregation and presentation during maturation of professional antigen presenting cells. PloS one. 2011;6:e16398.
- Ulbricht A, Gehlert S, Leciejewski B, et al. Induction and adaptation of chaperone-assisted selective autophagy CASA in response to resistance exercise in human skeletal muscle. Autophagy. 2015;11:538–546.
- Ghaoui R, Palmio J, Brewer J, et al. Mutations in HSPB8 causing a new phenotype of distal myopathy and motor neuropathy. Neurology. 2016;86:391–398.
- Cristofani R, Crippa V, Rusmini P, et al. Inhibition of retrograde transport modulates misfolded protein accumulation and clearance in motoneuron diseases. Autophagy. 2017;13:1280–1303.
- Rusmini P, Crippa V, Cristofani R, et al. The role of the protein quality control system in SBMA. J Mol Neurosci. 2016;58:348–364.
- Rusmini P, Cristofani R, Galbiati M, et al. The role of the Heat Shock Protein B8 (HSPB8) in motoneuron diseases. Front Mol Neurosci. 2017;10:176.
- Behl C. Breaking BAG: the co-chaperone BAG3 in health and disease. Trends Pharmacol Sci. 2016;37:672–688.
- Gamerdinger M, Kaya AM, Wolfrum U, et al. BAG3 mediates chaperone-based aggresome-targeting and selective autophagy ofmisfolded proteins. EMBO Rep. 2011;12:149–156.
- Gamerdinger M, Carra S, Behl C. Emerging roles of molecular chaperones and co-chaperones in selective autophagy: focus on BAG proteins. J Mol Med. 2011;89:1175–1182.
- Simeoni S, Mancini MA, Stenoien DL, et al. Motoneuronal cell death is not correlated with aggregate formation of androgen receptors containing an elongated polyglutamine tract. Hum Mol Genet. 2000;9:133–144.
- Piccioni F, Pinton P, Simeoni S, et al. Androgen receptor with elongated polyglutamine tract forms aggregates that alter axonal trafficking and mitochondrial distribution in motor neuronal processes. FASEB J. 2002;16:1418–1420.
- Poletti A. The polyglutamine tract of androgen receptor: from functions to dysfunctions in motor neurons. Front Neuroendocrinol. 2004;25:1–26.
- Rusmini P, Sau D, Crippa V, et al. Aggregation and proteasome: the case of elongated polyglutamine aggregation in spinal and bulbar muscular atrophy. Neurobiol Aging. 2007;28:1099–1111.
- Rusmini P, Bolzoni E, Crippa V, et al. Proteasomal and autophagic degradative activities in spinal and bulbar muscular atrophy. Neurobiol Dis. 2010;40:361–369.
- Rusmini P, Simonini F, Crippa V, et al. 17-AAG increases autophagic removal of mutant androgen receptor in spinal and bulbar muscular atrophy. Neurobiol Dis. 2011;41:83–95.
- Cristofani R, Crippa V, Vezzoli G, et al. The small heat shock protein B8 (HSPB8) efficiently removes aggregating species of dipeptides produced in C9ORF72-related neurodegenerative diseases. Cell Stress Chaperon. 2017. DOI:10.1007/s12192-017-0806-9.
- Crippa V, Sau D, Rusmini P, et al. The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS). Hum Mol Genet. 2010;19:3440–3456.
- Crippa V, Cicardi ME, Ramesh N, et al. The chaperone HSPB8 reduces the accumulation of truncated TDP-43 species in cells and protects against TDP-43-mediated toxicity. Hum Mol Genet. 2016;25:3908–3924.
- Cascella R, Fani G, Capitini C, et al. Quantitative assessment of the degradation of aggregated TDP-43 mediated by the ubiquitin proteasome system and macroautophagy. FASEB J. 2017. DOI:10.1096/fj.201700292RR.
- Zydowsky LD, Etzkorn FA, Chang HY, et al. Active site mutants of human cyclophilin A separate peptidyl-prolyl isomerase activity from cyclosporin A binding and calcineurin inhibition. Protein Sci. 1992;1:1092–1099.
- Aits S, Kricker J, Liu B, et al. Sensitive detection of lysosomal membrane permeabilization by lysosomal galectin puncta assay. Autophagy. 2015;11:1408–1424.
- Paz I, Sachse M, Dupont N, et al. Galectin-3, a marker for vacuole lysis by invasive pathogens. Cell Microbiol. 2010;12:530–544.
- Maejima I, Takahashi A, Omori H, et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. Embo J. 2013;32:2336–2347.
- Gudmand-Hoyer E, Fenger HJ, Skovbjerg H, et al. Trehalase deficiency in Greenland. Scand J Gastroenterol. 1988;23:775–778.
- Lin CH, Wu YR, Yang JM, et al. Novel lactulose and melibiose targeting autophagy to reduce PolyQ aggregation in cell models of spinocerebellar ataxia 3. CNS Neurol Disord-DR. 2016;15:351–359.
- Lee GC, Lin CH, Tao YC, et al. The potential of lactulose and melibiose, two novel trehalase-indigestible and autophagy-inducing disaccharides, for polyQ-mediated neurodegenerative disease treatment. Neurotoxicology. 2015;48:120–130.
- Rodriguez-Muela N, Hernandez-Pinto AM, Serrano-Puebla A, et al. Lysosomal membrane permeabilization and autophagy blockade contribute to photoreceptor cell death in a mouse model of retinitis pigmentosa. Cell Death Differ. 2015;22:476–487.
- Chauhan S, Kumar S, Jain A, et al. TRIMs and galectins globally cooperate and TRIM16 and galectin-3 co-direct autophagy in endomembrane damage homeostasis. Dev Cell. 2016;39:13–27.
- Kumar S, Chauhan S, Jain A, et al. Galectins and TRIMs directly interact and orchestrate autophagic response to endomembrane damage. Autophagy. 2017:1–2. DOI:10.1080/15548627.2017.1307487.
- Anding AL, Baehrecke EH. Cleaning house: selective autophagy of organelles. Dev Cell. 2017;41:10–22.
- Kawabata T, Yoshimori T. Beyond starvation: an update on the autophagic machinery and its functions. J Mol Cell Cardiol. 2016;95:2–10.
- Settembre C, Medina DL. TFEB and the CLEAR network. Methods Cell Biol. 2015;126:45–62.
- Zhang YJ, Xu YF, Cook C, et al. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci U.S.A. 2009;106:7607–7612.
- Crippa V, Carra S, Rusmini P, et al. A role of small heat shock protein B8 (HspB8) in the autophagic removal of misfolded proteins responsible for neurodegenerative diseases. Autophagy. 2010;6:958–960.
- Cashman NR, Durham HD, Blusztajn JK, et al. Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Devel Dyn. 1992;194:209–221.
- Durham HD, Dahrouge S, Cashman NR. Evaluation of the spinal cord neuron X neuroblastoma hybrid cell line NSC-34 as a model for neurotoxicity testing. Neurotoxicol. 1992;14:387–395.
- Reinhardt P, Glatza M, Hemmer K, et al. Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PloS one. 2013;8:e59252.
- Thellung S, Scoti B, Corsaro A, et al. Pharmacological activation of autophagy favors the clearing of intracellular aggregates of misfolded prion protein peptide to prevent neuronal death. Cell Death Dis. 2018;9:166.