
ABSTRACT
In the tumor stroma, cancer-associated fibroblasts (CAFs) affect all aspects of tumor evolution. Whereas several programs leading to CAF activation have been elucidated, little is known about the impact of the microenvironment on the turnover of key CAF regulators. RBPJ/CSL is a transcriptional repressor that mediates NOTCH signaling and its down-modulation activates the gene expression program(s) leading to stromal senescence and CAF activation. We overview our evidence that conditions increasing macroautophagy/autophagy, as often found in the stroma of tumors, cause the down-modulation of the RBPJ protein. This event requires the autophagic machinery and is functionally relevant because it is associated with an increase of CAF effector gene expression. The mechanism involves the direct association with the autophagy receptor SQSTM1/p62, which is required for RBPJ down-modulation. As a reflection of increased autophagy in the stroma, both the RBPJ and SQSTM1 proteins are down-modulated in Squamous Cell Carcinoma (SCC) patient-derived CAFs. Increasing RBPJ cellular levels stabilizes SQSTM1 and down-modulates the autophagic process. Our findings identify an autophagy-initiated mechanism for RBPJ down-modulation leading to increased CAF gene expression.
Although key for cancer initiation and progression, the involvement of autophagy in the stromal compartment, and in particular in cancer-associated fibroblast (CAF) conversion, is little investigated. Autophagy-activating conditions, such as low nutrients, increased ROS and hypoxic microenvironment, are often found in cancer stroma. The RBPJ/CSL protein, a transcriptional repressor converted by NOTCH into an activator, is key for negative control of the normal fibroblast conversion to CAF. Deletion of RBPJ in the mesenchymal skin compartment of mice or RBPJ down-modulation in primary human dermal fibroblasts (HDFs) results in the activation of a CAF-phenotype, which promotes and sustains the growth of neighboring cancer cells. Among RBPJ-regulated targets, the kinase ULK3 is responsible for CAF activation and concomitantly increases HDF autophagy, mitophagy and the associated metabolic reprogramming. So far little is known regarding the mechanism of RBPJ protein down-modulation and of the possible role of the microenvironment in this process. The autophagy receptor protein SQSTM1 (sequestosome 1) is a key cargo for the degradation of specific components, including transcription factors such as SMADs and NFKB/NF-ǀB, with SQSTM1 being itself degraded in the process. Importantly, the levels of SQSTM1 protein are downregulated in the stroma of several cancer types and is causative for the metabolic reprogramming of CAFs.
Figure 1. Control of RBPJ protein turnover in the tumor microenvironment. The transcriptional repressor that mediates NOTCH signaling, RBPJ, suppresses the gene expression program(s) leading from normal fibroblast to cancer-associated fibroblast (CAF) (left). RBPJ suppresses autophagy and mitophagy through transcriptional repression of the ULK3 gene, which, separately from these processes, causes CAF activation through GLI transcription factors. Conditions inducing autophagy, as often found in tumor stroma, cause the downregulation of RBPJ protein and concomitantly activate key CAF and autophagy effector genes. Mechanistically, RBPJ associates directly with the autophagy/signaling receptor SQSTM1, as part of a self-enforcing loop linking CAF activation with the autophagy process.
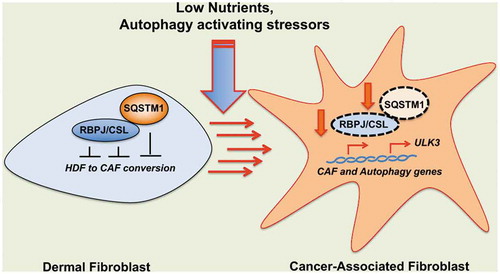
Previous evidence reported that pro-carcinogenic stimuli such as UVA and smoke extract exposure, which induce autophagy, can downregulate RBPJ in dermal fibroblasts. To understand the possible role of autophagy in down-modulation of this protein we exposed several HDFs strains to different experimental conditions activating different types of autophagy such as serum starvation, inhibition of MTOR activity or mitochondria uncoupling [Citation1]. We determined that all inducers have an impact on RBPJ protein levels; importantly the down-modulation of RBPJ protein levels is not a consequence of a reduced transcription because RBPJ mRNA is not concomitantly decreased.
Because loss of RBPJ in the stroma leads to CAF activation, we thus investigated whether the autophagy-induced RBPJ down-modulation is of functional significance, and we found that all activators of the autophagic process induce a simultaneous increase in transcription of key CAF effector genes such as PTGS2 (prostaglandin-endoperoxide synthase 2), TNC (tenascin C) and IL6 (interleukin 6).
To assess whether RBPJ protein down-modulation is indeed a consequence of increased autophagy, we employed several complementary approaches to inhibit the autophagic machinery. Pharmacologically, use of 3-methyladenine (3MA) to inhibit autophagy counteracts the down-modulation of RBPJ by various inducers of autophagy, while resulting in an concomitant accumulation of SQSTM1. Genetically, down-modulation of RBPJ by activators of autophagy is similarly suppressed in HDFs with silencing of the ATG5 gene and in mouse embryonic fibroblasts (MEFs) with Atg5 gene disruption.
Mechanistically, an attractive possibility is that the autophagy receptor protein SQSTM1 might mediate RBPJ degradation. By proximity ligation assay (PLA), which provides a sensitive method for detection of endogenous protein-protein association, we found puncta resulting from the close juxtaposition of anti-RBPJ and -SQSTM1 antibodies in HDFs. SQSTM1-RBPJ association was confirmed by co-immunoprecipitation of the endogenous proteins in HDFs and a direct interaction between the two was demonstrated by using the purified recombinant proteins. Of the several functional regions of SQSTM1 protein, which include an N-terminal oligomerization domain (PB1), a MAP1LC3/LC3 (microtubule associated protein 1 light chain 3) interacting motif (LIR), and a C-terminal ubiquitin-association domain (UBA), we mapped the RBPJ-binding region to the SQSTM1 C-terminal domain, which has been more directly implicated in the delivery of proteins for degradation.
We then assessed whether SQSTM1 is required for RBPJ degradation by autophagy and found that this is indeed the case, as down-modulation of RBPJ protein levels by pro-autophagic stimuli is suppressed in both HDFs or MEFs with siRNA- or shRNA- mediated Sqstm1 silencing. Similarly, RBPJ down-modulation is blocked in MEFs derived from mice with Sqstm1 gene disruption. These findings are of likely clinical significance for the conversion from HDFs to CAFs, in which RBPJ down-modulation induces autophagy. In fact, we found a parallel down-modulation of RBPJ and SQSTM1 levels in a set of skin Squamous Cell Carcinoma (SCC)-derived CAF strains versus normal HDF strains and, functionally, increased RBPJ expression results in concomitant SQSTM1 stabilization and suppression of autophagy (as assessed by expression of 3 autophagy markers genes, ULK3, MAP1LC3B and GABARAPL1). As decreased SQSTM1 can trigger CAF activation in multiple systems, stabilization of SQSTM1 by sustained RBPJ expression is consistent with the reversion of CAF activation that RBPJ overexpression can achieve.
Overall, our studies point to a novel role of the tumor microenvironment in controlling RBPJ protein levels in stromal fibroblasts, through an autophagy-dependent mechanism involving direct RBPJ-SQSTM1 protein association (). In turn, decreased RBPJ levels are required for induction of the autophagic process and concomitant downregulation of SQSTM1 protein levels. All the findings point to the existence of a self-reinforcing loop that could be targeted to interfere with CAF activation.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
Reference
- Goruppi S, Jo SH, Laszlo C, et al. Autophagy controls CSL/RBPJκ Stability through a p62/SQSTM1-dependent mechanism. Cell Rep. 2018 Sep 18;24(12):3108–3114.