
ABSTRACT
Macroautophagy/autophagy and innate immunity are central processes in neurodegeneration, but it has been unclear whether they work independently or in combination to assault the neuron. We recently demonstrated that reduced efficiency of autophagy causes hyperactivation of innate immunity, which in turn is necessary and sufficient for loss of dopaminergic neurons in a Cdk5-mediated model of degeneration in Drosophila. Genetically restoring autophagy, or reducing innate immune activation, rescues the dopaminergic neuron loss that occurs due to altered Cdk5 activity. This work revealed an alliance of innate immunity and autophagy that causes neuron loss in Cdk5-mediated neurodegeneration.
An intimate relationship exists between autophagy and neurodegeneration (ND), but the exact nature of that relationship remains mysterious. Is autophagy hyperactive in ND, or inactive, or is it active, but against the wrong targets? What is the connection between improper autophagy and other core processes of ND, such as innate immune response? Our studies of a Cdk5 (Cyclin-dependent kinase 5)-mediated model of neurodegeneration yielded 2 major results [Citation1]. First, hyperactive innate immunity is necessary and sufficient for neuron loss. Second, innate immune activation occurs as a consequence of reduced efficiency of autophagy (). Below, we describe the sequence linking these processes in Cdk5-mediated neurodegeneration.
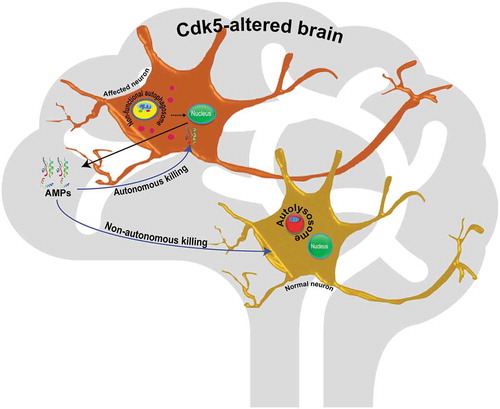
Figure 1. Schematic summarizing the link between autophagy and immunity in Cdk5-associated neurodegeneration. In affected neurons, inactive autophagosomes signal to the nucleus to enhance transcription of antimicrobial peptides, and these peptides either attack the expressing neuron autonomously, or attack nearby neurons, to induce cell death.
Hyperactive innate immunity induces neuron degeneration
The brain is largely considered to be immune privileged, but various evidence suggests a close, functional interaction of neuronal and immune systems. A major hurdle in investigating this relationship is the homeostatic nature of immune system regulation. Extreme enhancement or suppression of the innate immune response will undoubtedly impair the viability of the cell and of the organism, but not necessarily in the same way as in real disease. We therefore examined the status of innate immunity in our ND model, taking care to limit ourselves to modest modulation of immunity, within the range of changes that occur in the course of the natural disease.
We induced neurodegeneration in Drosophila by altering the activity of an atypical member of the cyclin dependent kinase family, Cdk5. Cdk5 is not activated by canonical cyclins; rather, its activity relies on binding to a regulatory subunit called Cdk5α (sometimes referred to as D-p35). Cdk5 activity is largely limited to postmitotic neurons, and changes in its activity are associated with various NDs such as Alzheimer and Parkinson. Either gain or loss of function of Cdk5 induces neuron death, both in vivo and in cell culture. Upon changing Cdk5 activity genetically in Drosophila, by altered expression of Cdk5α, we observe age-dependent degeneration of dopaminergic neurons. Prior to the onset of neuron loss, however, we also observe hyperactivation of the innate immune response, as assayed by global gene expression profiling. The innate immune response in the fly comprises several families of antimicrobial peptide (AMPs), along with other immune-inducible small proteins. Secretion of AMPs is tightly controlled by 2 different signaling pathways, known as Toll and Imd, and their associated NF\kappaB transcription factors, Dif, dl (dorsal) and Rel (Relish), and these pathways are conserved in the innate immune response of mammals.
The association of DA neuron degeneration with innate immune activation led us to investigate whether immune activation was the cause or consequence of cell loss. When individual AMPs are overexpressed in DA neurons using ple-GAL4, (a driver for dopaminergic neurons), age-dependent DA neuron loss is observed in otherwise wild-type flies. Conversely, reducing immune activation in flies with altered Cdk5 activity, by RNAi knockdown of Rel, or by a heterozygous mutant of Rel (RelE20), rescues DA neuron survival. Thus, activation of innate immunity is both necessary and sufficient to lead to Cdk5-associated loss of DA neurons in Drosophila.
Why does overexpression of innate immune components induce neuron loss? At least 3 possibilities present themselves. In the fly brain, AMP production is largely the job of circulating immune cells (sc-RNAseq database of Drosophila brain; http://scope.aertslab.org/). In the Cdk5 loss of function, however, we observe widespread production of AMPs by neurons. It may be that production of AMPs is intrinsically toxic to neurons, or that the AMP molecules made in neurons are somehow defective, and neurotoxic. Alternatively, it could simply be that the large number of neurons in the brain produces a toxic, high local concentration of AMPs. Further study will be required to test these hypotheses.
Reduced autophagy hyperactivates innate immunity to lead to degeneration of DA neurons
The traditional view of autophagy is that it maintains cellular homeostasis, in particular by clearing misfolded proteins and damaged cell organelles. That view has become more complicated, however, as autophagy has been found to be associated with the innate immune response and ND. Altered Cdk5 activity is associated with defective autophagy as well as with neurodegeneration. In the current work, we found that the link between these processes is mediated via innate immunity.
Microbial challenge is the primary stimulus activating innate immunity. However, we found no difference in bacterial load in animals with altered Cdk5 as they developed hyperactive immunity. Moreover, AMP overexpression is observed in degeneration-prone genotypes even in axenic conditions, ruling out the possibility that microbial exposure is responsible for immune hyperactivation in Cdk5-altered animals.
What then is inducing AMP overexpression? The first clue came from our expression profiling data, which revealed downregulation of various proteases and hydrolases in conjunction with overexpression of AMPs. Further study showed that this reflected decreased autophagy in Cdk5-altered flies. We found, for example, accumulation of ref(2)p/SQSTM1/p62, as well as evidence for failure of acidification of autolysosomes (assayed with a tandem tagged GFP-mCherry-Atg8 reporter), and reduced levels of various lysosomal proteins, including the Vha13 proton pump and the cysteine protease Cp1/Cathepsin L.
While these data clearly demonstrate impaired lysosomal metabolism and reduced autophagic efficiency, they still do not reveal the link of autophagy to innate immunity in Cdk5-altered animals. However, we found that reducing autophagy by mutation of 1 of the 2 Atg8 paralogs in the fly is sufficient to greatly increase expression of AMPs in otherwise wild-type flies. Conversely, gene expression data show that Mitf/TFEB, a master regulator of lysosomal metabolism, is downregulated in Drosophila lacking Cdk5 activity, and restoring autophagy to these flies by restoration of Mitf largely rescues AMP expression and DA neuron survival. Thus, it appears that reducing autophagy by itself is necessary and sufficient to induce immune activation. The link between autophagy and immune activation is further underlined by recent data that MITF also acts as a direct, negative regulator of immunity genes in mice. Thus, based on 3 independent lines of evidence, we know that reduced autophagy is necessary and sufficient for the innate immune activation that mediates neuron degeneration.
Conclusion
Overall, we revealed the relationship between autophagy and innate immunity in Cdk5-mediated neurodegeneration. Our data demonstrated that the reduced autophagy in these animals causes hyperactivation of innate immunity that leads to age-dependent loss of DA neurons, whereas restoration of autophagy is neuroprotective by restoring proper immunity. Considering the conservation of genes and signaling pathways between flies and mammals, these data suggest the hypothesis of a direct connection between autophagy and innate immunity in humans and open a new avenue for further exploration of the relationship between these 2 core processes in neurodegenerative disease.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Reference
- Shukla AK, Spurrier J, Kuzina I, et al. Hyperactive innate immunity causes degeneration of dopamine neurons upon altering activity of Cdk5. Cell Rep. 2019 Jan 2;26(1):131–144.e4. PMID: 30605670.