
ABSTRACT
Therapy resistance of tumor cells is a major obstacle for efficient anticancer treatment approaches and has been attributed to tumor heterogeneity as well as genetic and epigenetic changes. Accumulating evidence demonstrates that tumor cell adhesion to the extracellular matrix acts as an additional essential factor conferring tumor cell resistance to both radio- and chemotherapeutic intervention. Our recent study demonstrates that DDR1 (discoidin domain receptor tyrosine kinase 1) elicits therapy resistance of glioblastoma multiforme (GBM) stem-like and bulk cells through its adhesion to extracellular matrix and the subsequent modulation of macroautophagy/autophagy. Mechanistically, DDR1 associates with a YWHA/14–3-3-BECN1-AKT1 multiprotein complex favoring pro-survival/anti-autophagic and resistance-mediating AKT-MTOR signaling. In turn, inhibition of DDR1 sensitizes glioblastoma cells to radio- and chemotherapy by inducing autophagy. Collectively, our study suggests that DDR1 may be a potential target for sensitizing glioblastoma cells to combination therapies through its efficient induction of autophagic cell death.
Abbreviations
AKT1: AKT serine/threonine kinase 1; ATG14: autophagy related 14; BECN1: Beclin 1; DDR1: discoidin domain receptor tyrosine kinase 1; ECM: extracellular matrix; GBM: glioblastoma multiforme; MTOR: mechanistic target of rapamycin kinase; PDGFR: platelet derived growth factor receptor; PIK3C3: phosphatidylinositol 3-kinase catalytic subunit type 3; RPTOR: regulatory associated protein of MTOR complex 1; RICTOR: RPTOR independent companion of MTOR complex 2
The resistance of tumor cells to therapy frequently limits the efficiency of anticancer treatments. This may be intrinsic or acquired and ultimately causes poor prognosis and poor overall survival of patients suffering from cancer. In GBM, key factors driving such intrinsic or acquired therapy resistance include activating or repressive mutations and amplifications of pro-survival genes and epigenetic alterations. On top of these nuclear events, interactions of GBM cells with the extracellular matrix (ECM), especially collagens within the tumor microenvironment, have been defined in the past decade as additional critical determinants eliciting resistance mechanisms. With regard to the numerous types of cell adhesion molecules, efforts in targeting major integrin adhesion receptors, however, demonstrated poor efficacy to date. Our recent study focused on a particular type of cell adhesion molecule, termed DDR1 [Citation1]. We identified this receptor to be the only tyrosine-kinase containing collagen adhesion receptor overexpressed in GBM tumors compared to normal brain. We show that DDR1 is highly expressed in GBM stem-like cells and its colocalization with GBM stem cell markers in clinical samples associates with poor patient survival. While our data are in line with a reported prognostic value of DDR1 in other cancers, we additionally suggest a relevant, but unidentified, role of DDR1 for the highly therapy refractory GBM stem cell population, which is targeted in the clinic with multimodal treatment approaches.
We examined the unknown function of DDR1 for GBM therapy resistance using pharmacological DDR1 inhibition as well as siRNA-mediated DDR1 silencing in combination with clinical standard X-ray irradiation and temozolomide treatment. In both GBM stem-like cells and GBM cell lines, DDR1 deactivation reveals a promising radio- and chemosensitizing potential. Furthermore, blocking DDR1 in combination with radiochemotherapy in mice bearing orthotopic GBM results in tumor growth delay and prolonged survival of the animals. Collectively, our data suggest DDR1 as a potential molecular target for combinatorial GBM treatment approaches.
Our further explorations focused on untangling the molecular mechanism underlying therapy sensitization upon DDR1 inhibition. We applied broad-spectrum phosphoproteome analysis and found several DDR1-driven signaling hubs, such as the pro-survival AKT-MTOR and PDGFR pathway. These pathways are overactivated in GBM and mediate GBM tumorigenesis, cancer stem cell features and therapy resistance. By mass spectrometry analysis of the DDR1 interactome paired with DDR1 mutagenesis, we further identified YWHA/14–3-3 proteins as the linker between DDR1 and the AKT-MTOR signaling axis that acts by directly recruiting AKT1 (). Indeed, double silencing of DDR1 with either AKT1 or the MTOR-interacting molecules RPTOR or RICTOR shows the same degree of radiochemosensitization relative to single DDR1 silencing. These observations provide evidence for the concept of a jointly operated signaling mechanism involving DDR1, AKT1, and RPTOR-RICTOR.
Figure 1. Inhibition of DDR1 modulates radiosensitization through autophagy. Schematic representation of DDR1 function in GBM cells upon radiochemotherapy. Under basal conditions, DDR1 associates with YWHA/14–3-3-BECN1-AKT1 protein complex, mediates pro-survival DDR1 signaling and radiochemoresistance. Upon DDR1 inhibition, the YWHA/14–3-3-BECN1-AKT1 protein complex dissociates from DDR1. BECN1 interacts with PIK3C3/VPS34 and ATG14 and induces autophagic cell death forradiochemosensitization. P, phosphorylation.
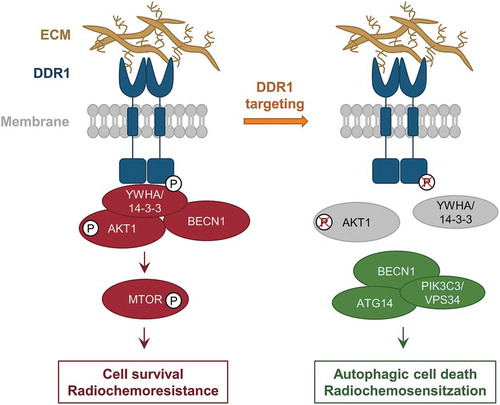
In addition, BECN1 co-precipitates with the DDR1-YWHA/14–3-3-AKT1 complex indicating a connection between the DDR1 adhesion complex and autophagy regulation (). Accordingly, inhibition and silencing of DDR1 enhances LC3-I-to-LC3-II conversion in GBM stem-like cells and orthotopic GBM tumors and increases the numbers of LC3-positive autophagosomes when combined with radiotherapy. Interestingly, apoptosis remains unaffected upon DDR1 inhibition. We provide further support for an induction of autophagy by discovering a disassembly of the DDR1-YWHA/14–3-3-BECN1-AKT1 complex upon DDR1 inhibition followed by a re-complexation of BECN1 with PIK3C3/VPS34 and ATG14, both proteins of the autophagy core complex (). To prove this concept further, BECN1 silencing results in an abrogation of the effects conferred by DDR1 inhibition on GBM cell survival upon irradiation. These data confirm that autophagy is causative for GBM therapy sensitization in the presence of deactivated DDR1.
In summary, our recent study exposes a so far unidentified DDR1-driven therapy resistance mechanism in GBM. Consequently, DDR1 inhibition may emerge as a promising therapeutic approach in a combination with radiochemotherapy in GBM patients. Additionally, our observations shed further light on how extracellular cues determine the pro-survival/autophagic tumor cell response to cytotoxic agents. In contrast to the notion of autophagic cell death counteracting anticancer treatment, our study reveals the potential of autophagic cell death induction for therapy sensitization, a circumstance to be explored in more depth in future studies.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Reference
- Vehlow A, Klapproth E, Jin S, et al. Interaction of discoidin domain receptor 1 with a 14-3-3-Beclin-1-Akt1 complex modulates glioblastoma therapy sensitivity. Cell Rep. 2019 Mar 26;26(13):3672–3683.e7.