
ABSTRACT
Intestinal epithelium functions as a barrier to protect the host from environmental microbes. Defects in macroautophagy/autophagy combined with intestinal microbes cause a disruption of homeostasis of the tissue, which is associated with the etiology of Crohn disease, an inflammatory bowel disease. However, the molecular mechanism of how autophagy interacts with microbes in the pathology are mostly unrevealed. Our recent findings using Drosophila as a model system showed that autophagy in enterocytes suppresses a regenerative response triggered by reactive oxygen species (ROS) secreted by the host epithelia toward commensal bacteria in the intestine. Without this suppression, accumulation of a receptor protein of selective autophagy, ref(2)P, continuously acts as a signaling platform to cause excessive regeneration via cytokine production by yki (yorkie) activation. This chronic response leads to the acceleration of age-dependent barrier dysfunction, systemic inflammation, and shorter lifespan. These results uncover a novel regulatory network linking commensal bacteria, autophagy, and gut homeostasis, represented by ROS, ref(2)P, and the hippo pathway.
Are commensal bacteria friends or foes? As many of the multicellular organisms live with bacteria on their surface, these bacteria are normally not harmful, or are even beneficial to the host. The intestine is a typical tissue in which bacteria are colonized, but also is often damaged by orally infected pathogenic bacteria. Against the harmful bacteria, the intestinal epithelium acts as a front line of host defense by activating immune systems. Conversely, the host tolerates symbiotic bacteria, and allows them to colonize in the intestinal tract. However, the host-microbe interactions are context dependent, and potentially symbiotic microbes often cause inflammation, although the underlying mechanism of which is poorly understood. Crohn disease (CD), a chronic disorder of the intestine, is characterized by progressive mucosal inflammatory conditions in the entire gastrointestinal tract. It has been revealed that the pathogenesis of CD relies on the interplay between genetic factors, such as autophagy factors ATG16L1 or IRGM, and environmental factors, such as enteric microbes. Although studies using a mouse model that harbors the risk allele of ATG16L1 have revealed the clear interaction of enteric microbes and the genetic risk with regard to the pathogenesis, it remains unrevealed if the effect of the interaction is causative or a consequence of the disease. One of the reasons for the difficulty in these analyses is because acquired immunity worsens the inflammation, even the etiology of which is innate immunity.
Drosophila has been widely used for studying intestinal homeostasis because of the easy manipulation of both the host genetic background and the gut microbes. In our study it is also useful because it has only innate immunity, but not acquired immunity for the defense against microbes. To test if the Drosophila model is useful for investigating the molecular mechanism underlying the etiology of CD, we first examined whether the defect of autophagy in enterocytes, the differentiated intestinal epithelial cells, causes similar phenotypes that are observed in the ATG16L1 model mouse intestine [Citation1]. The knockdown of the autophagy factors in enterocytes causes overproduction of cytokine upd3, that provokes over-proliferation of stem cells non-cell autonomously, resulting in a diffused pattern of septate junction proteins, such as dlg (discs large), in enterocytes, and the dysplasia of the epithelium (). Using flies with enterocyte-specific knockdown of Atg7, we next examined the molecular mechanism by which autophagy suppresses the disorder. It has been shown that in case of pathogenic bacteria infection, upd3 expression is activated by the transcription factor yki (yorkie; a homolog of mammalian YAP1), which is suppressed by the hippo pathway. Interestingly, a proteomic study has shown that dachs, an upstream regulator of the hippo pathway, makes a complex with the selective autophagy receptor protein ref(2)P, a mammalian SQSTM1/p62 homolog. Consistent with these studies and our findings that dachs and ref(2)P proteins do indeed physically interact with each other, we showed that dachs and ref(2)P proteins colocalize and act as a signaling platform at the apical side of the enterocyte to inactivate the hippo pathway, thereby activating yki-mediated upd3 expression. By recruiting ref(2)P protein to phagophores, these protein complex are quantitatively regulated by autophagy, which avoids the excessive proliferation of the stem cells. The trigger to cause the punctate structure of ref(2)P and dachs proteins is the microbicidal reactive oxygen species (ROS) that are produced by gut epithelia. To our surprise, it is not pathogenic bacteria, but nonpathogenic commensal bacteria, which provoke no chronic responses in healthy intestine, that cause the disorder in the intestine with defective autophagy. Importantly, the excessive proliferation of the stem cells in the presence of symbiotic bacteria in autophagy-defective intestine results in a chronic regenerative response, that leads to acceleration of age-related barrier dysfunction, systemic inflammation and shorter lifespan. Therefore, autophagic regulation of the hippo pathway via ref(2)P–dachs clearance is an essential mechanism to tolerate symbiotic bacteria in the intestinal tract for a lifetime.
Figure 1. Autophagy in Drosophila enterocytes regulates regenerative responses in response to enteric bacteria. Host enterocytes secrete microbicidal ROS toward intestinal flora, which in turn stimulate the regenerative signaling in enterocytes via oligomerization of the ref(2)P protein. Top left: In a homeostatic condition, autophagy degrades ref(2)P oligomers together with dachs protein, an upstream regulator of the hippo pathway, to suppress excessive regeneration. Top right: If the autophagic activity is impaired by genetic mutation and/or organismal aging, the oligomeric structure of ref(2)P accumulates in enterocytes, resulting in the over-proliferation of intestinal stem cells. Bottom: Because host enterocytes enhance ROS secretion upon pathogenic infection, larger numbers of ref(2)P oligomers are formed under this condition. Therefore, it is plausible that the increased number of ref(2)P oligomers can overcome the autophagic regulation and activate regenerative responses to compensate for the loss of damaged cells
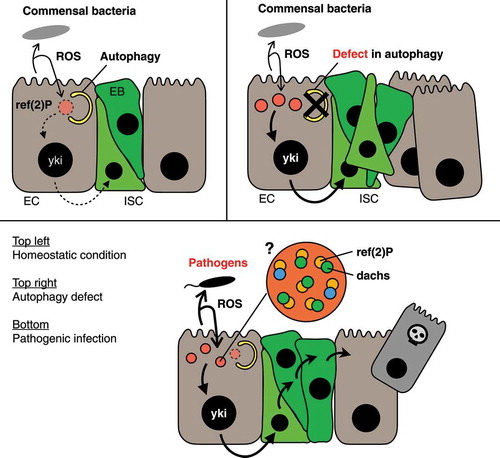
Our study uncovered the linkage between autophagic regulation of the hippo pathway and the mechanism of keeping the homeostatic state of the gut with symbionts. Given the functional conservation of ref(2)P and SQSTM1, it is of interest to know if SQSTM1 mediates any signaling in the mammalian intestine, which is regulated by selective autophagy. Our findings also raise an interesting question of how ref(2)P protein forms a punctate structure with a diameter from 0.6 to 2.5 µm in response to ROS stimulation. Because recent studies have revealed that mammalian SQSTM1 forms a round structure of similar size by liquid-liquid phase separation, and colocalizes with its binding partner, it is possible that sensing ROS damage by an unknown factor(s) may enable ref(2)P and dachs proteins to condense at the formation site of the structure, which triggers the phase separation. In this context, it is interesting that we observed the ref(2)P structure at the sub-apical region of the enterocytes, because in mammalian epithelial cells, TJP/zona occludens (tight junction protein) proteins, scaffolding proteins required for tight-junction assembly, are phase-separated at sub-apical region, and colocalize with multiple proteins including YAP1. Further studies of the mechanism of ROS–dependent ref(2)P structure formation, as well as the studies of whether SQSTM1 has any function on regulating signaling pathways in the mammalian intestine, will open the door for understanding the unrevealed etiology of inflammatory bowel disease.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Reference
- Nagai H, Tatara H, Tanaka–Furuhashi K, et al. Homeostatic regulation of ROS-triggered Hippo-Yki pathway via autophagic clearance of Ref(2)P/p62 in the Drosophila intestine. Dev Cell. 2021;56(1):81–94. PMID: 33400912. .