
ABSTRACT
Alzheimer disease (AD) is a neurodegenerative disorder for which no approved medication exists. AD is characterized by worsening cognitive and non-cognitive symptoms, and research in the AD field strives to identify very precocious brain alterations leading to an irreversible condition. Recently it has been demonstrated that several early AD symptoms are paralleled with degeneration of neurons producing dopamine (DA), a neurotransmitter involved in the regulation of cognitive and non-cognitive functions. Actually, we found that ventral tegmental area (VTA) DA neurons degenerate early in a validated AD mouse model (Tg2576). Here, we summarize new data showing how macroautophagy/autophagy impairment – due to enhanced activity of the ABL/c-Abl kinase – might cause the DA neuron loss. We also proved that nilotinib, an ABL inhibitor, restores autophagy flux, thus preventing VTA neurodegeneration. Most notably, from a clinical point of view, nilotinib, by preventing DA neuronal loss, preserves DA outflow in VTA-projecting areas, improving Tg2576 behavioral phenotypes. Our findings shed light on the mechanism involved in DA neurodegeneration, revealing that autophagy represents a viable therapeutic target in early AD.
The VTA is the origin of axons forming the mesocorticolimbic dopaminergic pathway, which regulates several functions, including cognition and motivation, by targeting the hippocampus, prefrontal cortex, nucleus accumbens (NAc) and amygdala. The relevance of DA for hippocampal-dependent memory and plasticity and for NAc-related reward circuitry led to multiple studies proving a link between AD and deficits in mesocorticolimbic DA signaling both in experimental models and patients.
We had previously shown that an early, progressive, and selective loss of VTA DA neurons in Tg2576 mice occurs before Aβ plaque deposition, leading to low DA levels in the hippocampus and NAc, and causing memory and reward deficits, respectively. Moreover, independent clinical studies in mild cognitive impairment due to AD and AD patients described functional, structural, and metabolic changes in the VTA or its projection areas, highlighting the VTA volume or connectivity as clinical markers for conversion from healthy state to clinical AD.
However, although the interest in this topic is growing rapidly and several lines of evidence suggest that VTA DA neurons display high vulnerability – due to their intrinsic properties – the molecular mechanisms involved in DA neuron loss in AD are still unknown. Curiously, autophagy seems to play an important role in these neurons as proved by its high levels in physiological conditions.
In our work we investigated in Tg2576 mice whether impairment in the autophagic machinery is involved in VTA DA neuron loss at the beginning of neurodegeneration [Citation1]. Particularly, by electron and confocal microscopy we demonstrated an increased autophagosome density in DA neurons from 3-month-old Tg2576 mice, suggesting an alteration in the autophagic mechanism. Autophagy is a highly dynamic, multi-step process, and autophagosome accumulation could occur due to increased formation of autophagic vesicles or to decreased fusion with lysosomes (to form autolysosomes). To discriminate between these events, we used an adeno-associated virus carrying the fluorescent reporter mCherry-GFP-LC3. Based on the differential pH sensitivity of mCherry and GFP, we quantified autophagosomes and autolysosomes demonstrating that the autophagy flux impairment was due to increased autophagosome production.
Interestingly, autophagy is a critical regulator of neuronal activity and the increased spontaneous firing frequency that we recorded in VTA DA neurons at 3 months – that becomes more severe at 6 months – could be associated with autophagy flux alterations. The observed neuronal shrinkage could also be considered as a parameter linked with the disease progression ().
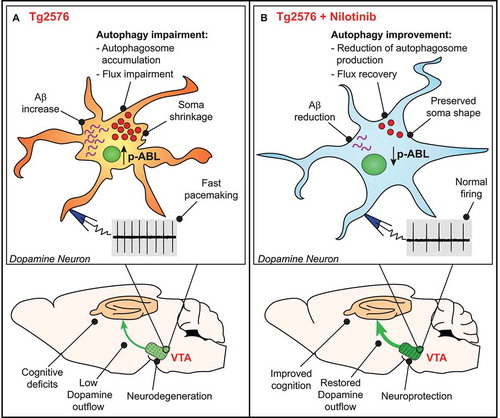
Figure 1. Nilotinib exerts neuroprotective effect on VTA dopamine neurons by the improvement of autophagic flux. (A) VTA dopamine neurons from 3-months-old Tg2576 mice show increased phosphorylated ABL levels and impairment in autophagy machinery, which could cause accumulation of intracellular Aβ and increase in spontaneous firing frequency. During the disease progression, firing frequency alteration become more severe and cell shrinkage occurs. All these physiological alterations are strictly related with VTA neurodegeneration, which leads to a reduction of DA levels in VTA-projecting areas, such as the hippocampus, and cognitive deficits. (B) VTA DA neurons from nilotinib-treated mice show reduction of both ABL phosphorylated levels and autophagosomes accumulation. The improvement in autophagy efficiency promotes the reduction of Aβ and improves neuronal health, as demonstrated by normal firing frequency and preserved soma shape. Nilotinib exerts a neuroprotective effect on VTA DA neurons, thus preserving DA outflow in hippocampus and ameliorating memory functions in Tg2576 mice
To better characterize the autophagic deficit in Tg2576 DA neurons, we focused on a key regulator of this process – particularly studied in neurodegenerative diseases – the ABL tyrosine kinase. We found a selective increase of ABL phosphorylation in the Tg2576 midbrain, which suggests an increased activity of ABL at the beginning of VTA neurodegeneration (). Thus, to reduce ABL activity, we performed a chronic treatment with nilotinib, an ABL inhibitor approved by the U.S. FDA for adults with chronic myeloid leukemia. Our interest in this drug is due to results obtained in AD and Parkinson disease (PD) animal models, in which nilotinib induces the autophagic degradation of Aβ and SNCA/α-synuclein, improving cognitive and motor performances, respectively. Moreover, clinical trials on AD and PD patients showed that nilotinib reduces disease-related biomarkers. Of note, we delivered the pharmacological treatment before VTA neurodegeneration. Nilotinib reduced ABL phosphorylation and improved autophagic clearance, by reducing autophagosome accumulation in DA neurons and restoring autophagy flux. Because autophagy impairment is related to protein accumulation, we investigated the effect of nilotinib on Aβ levels in brain areas involved in AD-related symptoms. Consistent with previous observations, nilotinib promotes Aβ clearance not only in VTA DA neurons, but also in several brain regions, suggesting a global effect of the treatment ().
Overall, the main goal of our work was to demonstrate that a chronic treatment targeting ABL prevents VTA DA neurodegeneration in Tg2576 and this result is probably due to the ability of nilotinib to improve autophagy. As a functional read-out of neuronal survival, nilotinib-treated Tg2576 mice show reduced spontaneous firing frequency, suggesting that the recovery of the autophagy process has an impact on physiological properties. The neuroprotective effect played by nilotinib on VTA DA neurons leads to the preservation of DA levels in the hippocampus and to significant improvements of memory-related performances in treated mice compared to controls ().
We are aware that this study does not dissect all molecular mechanisms linking ABL phosphorylation with the autophagy machinery, and this is mainly due to the functional complexity and heterogeneity of VTA neurons. However, for the first time we identified very early pathological changes in VTA DA neurons, which should be considered as prodromal markers to be validated in the human disease. Moreover, until now, autophagy impairments in AD have always been investigated in association with Aβ plaque accumulation rather than focusing on its physiological role in neuron survival. As mentioned, DA neurons have high physiological levels of autophagy, and its alteration – in combination with the peculiar activities of these cells – makes them particularly vulnerable compared to other neurons.
Collectively, our findings open to new possible clinical therapeutic strategies to target the mesocorticolimbic system in prodromal or early-diagnosed mild cognitive impairment, to delay the disease progression.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Reference
- La Barbera L, Vedele F, Nobili A, et al. Nilotinib restores memory function by preventing dopaminergic neuron degeneration in a mouse model of Alzheimer’s disease. Prog Neurobiol. 2021;102031. DOI:10.1016/j.pneurobio.2021.102031