
ABSTRACT
Macroautophagy/autophagy-related proteins regulate infectious and inflammatory diseases in autophagy-dependent or -independent manner. However, the role of a newly identified mammalian-specific autophagy protein-BECN2 (beclin 2) in innate immune regulation is largely unknown. Here we showed that loss of BECN2 enhanced the activities of NLRP3, AIM2, NLRP1, and NLRC4 inflammasomes upon ligand stimulations. Mechanistically, BECN2 interacted with inflammasome sensors and mediated their degradation through a ULK1- and ATG9A-dependent, but BECN1-WIPI2-ATG16L1-LC3-independent, non-canonical autophagic pathway. BECN2 recruited inflammasome sensors on ATG9A+ vesicles to form a complex (BECN2-ATG9A-sensors) upon ULK1 activation. Three soluble NSF attachment protein receptor (SNARE) proteins (SEC22A, STX5, and STX6) were further shown to mediate the BECN2-ATG9A-dependent inflammasome sensor degradation. Loss of BECN2 promoted alum-induced peritonitis, which could be rescued by the ablation of CASP1 in Becn2-deficient mice. Hence, BECN2 negatively regulated inflammasome activation to control inflammation, serving as a potential therapeutic target for the treatment of infectious and inflammatory diseases.
Abbreviations: AIM2: absent in melanoma 2; ATG: autophagy related; BECN1: beclin 1; BMDC: bone marrow-derived dendritic cells; BMDM: bone marrow-derived macrophages; CASP1: caspase 1; CQ: chloroquine; gMDSC: granulocytic myeloid-derived suppressor cells; IL: interleukin; LPS: lipopolysaccharide; MAP1LC3B: microtubule associated protein 1 light chain 3 beta; mMDSC: monocytic myeloid-derived suppressor cells; NLRC4: NLR family CARD domain containing 4; NLRP1: NLR family pyrin domain containing 1; NLRP3: NLR family pyrin domain containing 3; PECs: peritoneal exudate cells; PYCARD/ASC: apoptosis-associated speck-like protein containing a caspase activation and recruitment domain; SNAREs: soluble NSF attachment protein receptors; STX5: syntaxin 5; STX6: syntaxin 6; ULK1: unc-51 like autophagy activating kinase 1; WIPI: WD repeat domain, phosphoinositide interacting.
Introduction
Macroautophagy/autophagy-related proteins play an important role in infectious, autoimmune, and inflammatory diseases by regulating the innate immune signaling such as inflammasome and type I interferon [Citation1–5]. Autophagy protein has been reported to regulate the inflammasome activation through autophagic mechanisms, including the removal of intracellular inflammasome-activating danger/damage-associated molecular patterns, the sequestration and degradation of inflammasome components, and the control of biogenesis and secretion of IL1B/IL-1β (interleukin 1 beta) protein [Citation1–5]. Emerging evidence also shows that autophagy-related proteins could function in autophagy-independent or non-canonical autophagic pathways such as vesicular trafficking, innate immunity, cell death and proliferation [Citation3,Citation6–10]. BECN2 (beclin 2) has recently been reported to degrade several G protein-coupled receptors through an endo-lysosomal degradation pathway to control the obesity, insulin resistance, and the development of herpesvirus-associated Kaposi sarcoma [Citation11,Citation12]. In our recent study, we found that mice with homozygous ablation of becn2 produced higher amounts of proinflammatory cytokines including IL6 and IL1B, and had a markedly increased incidence of lymphoma development [Citation13]. BECN2 functions as a negative regulator in the control of the MAPK1/ERK2-MAPK3/ERK1 and NFKB/NF-κB signaling pathways to suppress IL6-mediated inflammation and spontaneous lymphoma development [Citation13]. However, its role in inflammasome activation and IL1B production remains largely unknown.
Inflammasome pathways play crucial roles in the host defense against invading pathogens, cancer development, metabolic diseases, and neurodegenerative diseases [Citation14–16]. Inflammasomes are multiprotein complexes that consist of a sensor protein, an adaptor protein PYCARD/ASC (apoptosis-associated speck-like protein containing a caspase activation and recruitment domain), and the pro-CASP1 (caspase 1) protein [Citation17–21]. Several established inflammasome complexes are composed of different NOD-like receptors, including NLRP3 (sensors for particulates, ATP and nigericin), AIM2 (sensors for dsDNA from microbes or host origin), NLRC4 (sensor for bacterial flagellin), and NLRP1 (sensor for Bacillus anthracis lethal toxin, Toxoplasma gondii, and muramyl dipeptide) [Citation17–21]. Ligand stimulation of these sensors leads to the recruitment and assembly of the inflammasome complex to activate CASP1 enzyme activity [Citation18,Citation19,Citation21]. The processed and active CASP1 further catalyzes the proteolytic cleavage of pro-IL1B and pro-IL18 to release mature IL1B and IL18 proteins [Citation17–21]. Various regulatory proteins have been identified to control the activation of inflammasomes, including GBP5, NEK7, EIF2AK2/PKR, BRCC3, and autophagy proteins, but their molecular mechanisms remain to be defined [Citation1–4,Citation22–25]. In this study, we showed that BECN2 regulated inflammasome activation by interacting with NLRP3, AIM2, NLRC4, and NLRP1 and targeting them for degradation through a ULK1- and ATG9A-dependent, but ATG7-, ATG16L1-, LC3-, WIPI2-, and BECN1-independent, non-canonical autophagic pathway. Importantly, the soluble NSF attachment protein receptors (SNAREs), including STX5, STX6, and SEC22A, were required for the BECN2-ATG9A-dependent degradation of inflammasome sensor proteins. Therefore, our findings have identified a previously unrecognized role of BECN2 in the negative regulation of inflammasome sensor proteins through ULK1- and ATG9A-dependent non-canonical autophagy, thus providing potential therapeutic targets for the treatment of inflammatory diseases.
Results
BECN2 deficiency enhances inflammasome activation
To determine the potential role and involvement of BECN2 in the inflammasome pathway, we evaluated the mRNA level of Becn2 in different organs and cell types from WT mice. We found that Becn2 was highly expressed in brain, spleen, testis, bone marrow-derived macrophages (BMDMs) and bone marrow-derived dendritic cells (BMDCs), especially in the thymus, T cells and B cells (Fig. S1A), indicating a potential function in innate and adaptive immune systems. To test the functional relevance of BECN2 in inflammasome activation, we stably expressed mCherry-tagged BECN2 in THP-1 cells by retrovirus transduction (Fig. S1B), and found that the IL1B production and cleaved CASP1 levels were markedly reduced in mCherry-BECN2 transduced THP-1 cells compared to mCherry-empty vector (EV) transduced control cells, in response to the stimulation from multiple ligands including nigericin (NLRP3 ligand), poly(dA:dT) (AIM2 ligand), anthrax lethal factor (LF, NLRP1 ligand), and flagellin (NLRC4 ligand) ( and ). Similar results were observed in BECN2-expressing HEK293T-CIA cells (293 T cells stably expressing CASP1, pro-IL1B, and PYCARD) [Citation26]. After transfecting plasmids encoding AIM2 or NLRP3 and stimulating the cells with poly(dA:dT) or nigericin, respectively, we showed that the IL1B production and cleaved CASP1 levels in the supernatant were markedly reduced in the cells co-transfected with increasing amounts of BECN2 expression plasmid DNAs in a dose-dependent manner (Fig. S1C-F). Consistently, knockout (KO) of BECN2 in THP-1 cells significantly enhanced the IL1B production and CASP1 cleavage under the treatment of nigericin, poly(dA: dT), LF, or flagellin ( and ).
Figure 1. BECN2 negatively regulates inflammasome activation and IL1B production. (A and B) PMA pre-treated mCherry-EV and mCherry-BECN2 overexpressed THP-1 cells were primed with LPS (200 ng/ml) for 3 h, and then stimulated with nigericin (1 μM, 6 h), poly(dA:dT) (1 μg/ml, 6 h), anthrax lethal factor (LF) (1 μg/ml, 4–6 h), or flagellin (20 μg/ml, 6–8 h), respectively. IL1B production in supernatants (SN) was examined by ELISA (A). Protein levels of pro-CASP1 in whole cell lysates (WCL) and cleavage CASP1 in SN were detected using immunoblotting (B). (C and D) PMA pre-treated WT and BECN2 KO THP-1 cells were primed with LPS (200 ng/ml) for 3 h, and then stimulated with nigericin (1 μM, 6 h), poly(dA:dT) (1 μg/ml, 6 h), anthrax lethal factor (LF) (1 μg/ml, 4–6 h), flagellin (20 μg/ml, 6–8 h). IL1B production in SN was examined by ELISA (C). Protein levels of pro-CASP1 in WCL and cleavage of CASP1 in SN were detected using immunoblotting (D). WT and Becn2-deficient mouse BMDM (E and F), peritoneal neutrophils (G and H), and bone marrow-derived dendritic cells (BMDC) (I and J) were primed with LPS (100 ng/ml, 3 h), and then stimulated with nigericin (1 μM, 6 h), poly(dA:dT) (1 μg/ml, 6 h), anthrax lethal factor (LF) (1 μg/ml, 4–6 h), flagellin (20 μg/ml, 6–8 h). IL1B production in SN was examined by ELISA (E, G, I). Protein levels of pro-CASP1 in WCL and cleaved CASP1 in supernatants (SN) were detected using immunoblotting (F, H, J). *P < 0.05, **P < 0.01, ***P < 0.001. (Student’s t-test). Data are plotted as mean ± s.e.m. of three independent experiments.
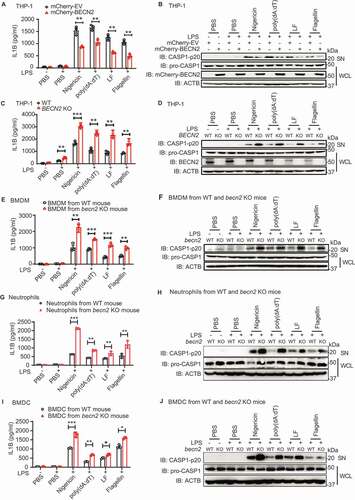
To further substantiate these findings under physiological conditions, we used the homozygous becn2 KO mice with validation of the allele deletion by polymerase chain reaction (PCR) (Fig. S1G). Because BECN2 is highly expressed in innate immune cells [Citation13], we isolated BMDM ( and ), neutrophils ( and ), and BMDC ( and ) from WT and becn2 KO mice and treated them with lipopolysaccharide (LPS), followed by different ligand stimulation. We found that all three types of cells obtained from becn2 KO mice produced significantly higher amounts of IL1B and cleaved CASP1 than those from WT counterparts, suggesting that BECN2 negatively regulates the inflammasome activation in multiple innate immune cells. Since IL1B is generated from the proteolytic cleavage of its precursor protein by active CASP1, the level of pro-IL1B is also critical for the production of mature IL1B. We determined the levels of pro-IL1B in LPS-primed neutrophils and BMDM, and found that pro-IL1B in becn2 KO cells was significantly higher than that in WT cells (Fig. S1H). By contrast, pro-IL1B level was comparable in WT and becn2 KO BMDC (Fig. S1H). Consistently, we showed that after LPS treatment, becn2 KO mice produced more IL1B in the serum, compared with WT mice (Fig. S1I). Together, these data suggest that the loss of BECN2 markedly enhanced the inflammasome activities upon ligand stimulation, but how BECN2 modulates the inflammasome signaling pathway remains unclear.
BECN2 interacts with inflammasome sensors via CCD-ECD domains
To determine how BECN2 negatively regulates inflammasome activation, we investigated whether BECN2 interacted with protein components (NLRP3, AIM2, NLRP1, NLRC4, PYCARD and CASP1) in the inflammasome complexes. Co-immunoprecipitation and western blot analyses using 293 T cells demonstrated that BECN2 could interact with NLRP3, AIM2, NLRP1, NLRC4 and CASP1, but not with PYCARD (). We further used THP-1 cells to show endogenous interaction between BECN2 and AIM2 or NLRP3, and found that such interactions were increased after poly(dA:dT) or nigericin treatment, respectively ( and ). Consistent with the endogenous interactions, confocal microscopic analysis demonstrated the colocalization of GFP-BECN2 with AIM2, or with NLRP3, in the cytosol of HeLa cells ( and ). These results clearly suggest that BECN2 was capable of interacting with inflammasome sensors under physiological conditions.
Figure 2. BECN2 interacts with inflammasome sensors through its CCDECD domain. (A) HEK293T cells were co-transfected with HA-tagged BECN2 and Flag-tagged EV, AIM2, NLRP1, NLRP3, NLRC4, PYCARD or CASP1. Cell lysates were immunoprecipitated with anti-Flag beads and immunoblotted using indicated antibodies. (B and C) THP-1 cells were left untreated or primed with LPS, then stimulated with poly(dA:dT) (1 μg/ml, 6 h) (B), or nigericin (1 μM, 6 h) (C). Cell lysates were immunoprecipitated using anti-NLRP3 or anti-AIM2 antibody and protein A/G beads and then immunoblotted using indicated antibodies. (D and E) HeLa cells co-transfected with GFP-BECN2 and Flag-AIM2 (D) or Flag-NLRP3 (E) were fixed and stained with anti-Flag antibody and the secondary antibody conjugated to Alexa Fluor 633. Colocalization was observed under confocal microscopy. Scale bar: 5 μm. (F) Schematic diagram of NLRP3 and domain truncation constructs. (G) HEK293T cells were co-transfected with Flag-tagged BECN2 and HA-tagged NLRP3 full-length or NLRP3 domain truncations. Cell lysates were immunoprecipitated with anti-Flag beads and immunoblotted using indicated antibodies. PYD, pyrin domain; LRR, leucine-rich repeat motif; NACHT, NOD or NBD-nucleotide-binding domain. (H) Schematic diagram of AIM2 and domain truncation constructs. (I) HEK293T cells were co-transfected with HA-tagged BECN2 and Flag-tagged AIM2 full-length or AIM2 domains, then immunoprecipitated using anti-Flag beads and immunoblotted using indicated antibodies. (J) Schematic diagram of BECN2 and domain truncation constructs. N, N-terminal domain; BH3 domain, BCL2 binding domain; CCD, central coiled-coil domain; and ECD, C-terminal evolutionarily conserved domain. (K and L) HA-tagged BECN2 full-length or domains were co-transfected with Flag-tagged AIM2 (K) or NLRP3 (L), followed by immunoprecipitation using anti-Flag beads and immunoblotted using indicated antibodies.
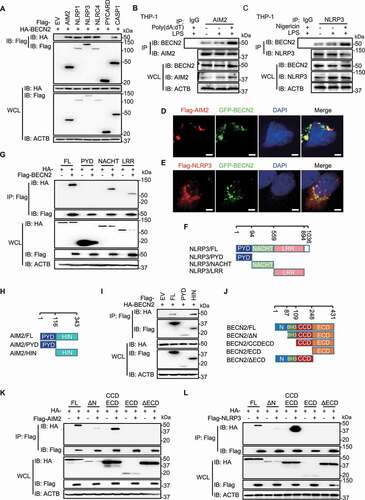
To further determine which domains of AIM2 and NLRP3 are responsible for their interactions with BECN2, we generated a series of truncated constructs (NLRP3-PYD, NLRP3-NACHT, NLRP3-LRR, AIM2-PYD, and AIM2-HIN domains) and found that the HIN domain of AIM2, as well as the NACTH and LRR domains of NLRP3, were responsible for their specific interactions with BECN2 (-). To identify which domain of BECN2 is required for its interaction with AIM2 and NLRP3, we generated four truncated BECN2 constructs, including N-terminal deletion fragment (BECN2/ΔN), CCD and ECD domains (BECN2/CCDECD), ECD domain alone, and ECD domain deletion fragment (BECN2/ΔECD) (). The co-immunoprecipitation of these truncations with AIM2 or NLRP3 revealed that the BECN2 CCD-ECD domains, but not other domains, interacted with AIM2 and NLRP3 ( and ). To determine whether the interaction between BECN2 and inflammasome sensors would affect the assembling of inflammasome complexes, HEK293T cells were co-transfected with HA-PYCARD and HA-CASP1 along with Flag-AIM2 or Flag-NLRP3 in the control or BECN2-expressing 293 T cells. We showed that the assembly of inflammasome components was not affected by BECN2 ectopic expression, as determined by co-immunoprecipitation assay (Fig. S2A and S2B), suggesting that BECN2 interacted with inflammasome sensors (AIM2 or NLRP3), but did not disrupt inflammasome assembly.
BECN2 targets inflammasome sensors for degradation through a lysosomal pathway
To understand how BECN2 inhibits inflammasome activation, we reasoned that BECN2 might target inflammasome sensors for degradation, since BECN2 has been reported to degrade G-protein coupled receptors through a lysosomal degradation pathway [Citation11]. We transfected 293T cells with key inflammasome components along with increasing amounts of BECN2 plasmids, and found that the protein levels of inflammasome sensors (AIM2, NLRP3, NLRP1, and NLRC4), but not PYCARD or CASP1, were markedly reduced with increasing BECN2 expression ( and S2C). Consistently, the endogenous inflammasome sensors, but not PYCARD, were also markedly decreased in mCherry-BECN2-expressing THP-1 cells, compared with mCherry-EV control-transduced cells ().
Figure 3. BECN2 degrades inflammasome sensors through the lysosomal pathway. (A) HEK293T cells were co-transfected with increasing doses of HA-tagged BECN2 plasmids along with Flag-tagged AIM2 or NLRP3, then immunoblotted using indicated antibodies. (B-D) WT and mCherry-BECN2-overexpressing THP-1 cells (B), or BECN2 KO THP-1 cells (C), and BMDMs from WT and Becn2-deficient mice (D) were left untreated or primed with LPS (200 ng/ml) for 3 h, then the protein levels of inflammasome sensors were detected by immunoblot using indicated antibodies. (E and F) HEK293T cells were co-transfected with HA-tagged BECN2 and Flag-tagged AIM2 (E) or NLRP3 (F), then treated with DMSO (vehicle), MG132, chloroquine (CQ) and bafilomycin A1 (Baf A1) for 6 h. Cell lysates were then immunoblotted using indicated antibodies. Quantification analysis for B-F is calculated based on the band density of three independent experiments after normalization. Statistical differences among inhibitor-treated groups were calculated using 1-way ANOVA with Tukey’s multiple comparison test (E, F). *P < 0.05, **P < 0.01, ***P < 0.001.
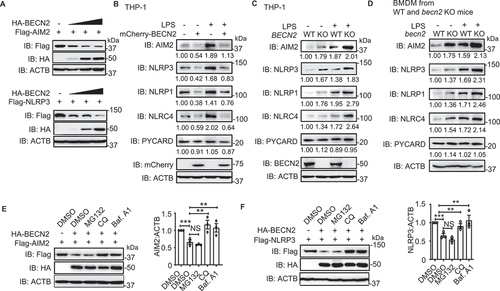
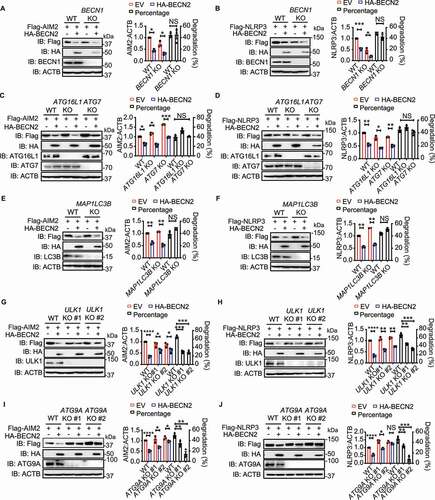
To provide the direct evidence that BECN2 regulates the stability of inflammasome sensors under physiological conditions, we generated BECN2-deficient THP-1 cells using CRISPR/Cas9 technology and found that the protein levels of inflammasome sensors (AIM2, NLRP3, NLRP1, and NLRC4) in BECN2-deficient THP-1 cells were higher than those in WT THP-1 cells with or without LPS treatment (). To further substantiate these findings, we isolated macrophages from WT and becn2 KO mice, and consistently found that the protein levels of inflammasome sensors (AIM2, NLRP3, NLRP1, and NLRC4) in macrophages derived from Becn2-deficient mice were markedly higher than those from WT mice, suggesting that BECN2 negatively regulates inflammasome sensors at the protein level in vivo (). By contrast, no appreciable difference in PYCARD protein level was found between macrophages isolated from WT and becn2 KO mice (). Furthermore, we showed no appreciable difference at the mRNA levels of AIM2 and NLRP3 between WT and Becn2-deficient mouse BMDMs or human THP-1 cells (Fig. S2D and S2E). These results suggest that BECN2 negatively regulated AIM2 and NLRP3 proteins at the post-transcriptional level. To determine the protein degradation kinetics of AIM2 and NLRP3, we performed a cycloheximide (CHX, protein synthesis inhibitor) “chase” assay in WT and BECN2 KO 293T cells. BECN2 KO 293T cell clones were generated by CRISPR/Cas9 technology and identified by sequencing of gDNA (Fig. S2F). We found that the deficiency of BECN2 in HEK293T cells markedly prolonged the half-life of AIM2 and NLRP3 (Fig. S2G), suggesting that BECN2 mediates rapid degradation of AIM2 and NLRP3.
To identify the functional domain of BECN2 in mediating the degradation of inflammasome sensors, we co-transfected Flag-AIM2 or Flag-NLRP3 along with different HA-BECN2 domains, respectively. The immunoblotting analysis revealed that the BECN2 CCD-ECD domain, which has been identified to interact with AIM2 and NLRP3 ( and ), was also required for the degradation of AIM2 and NLRP3 (Fig. S2H and 2I). Since there are two major (proteasomal and lysosomal) pathways for protein degradation, we next sought to determine which pathway is required for BECN2-mediated degradation of inflammasome sensors. We treated cells with lysosomal inhibitors such as bafilomycin A (BafA) and chloroquine (CQ) or proteasome inhibitor (MG132), and found that the degradation of AIM2 and NLRP3 was completely blocked by the lysosomal inhibitors (BafA and CQ), but not by the proteasomal inhibitor MG132 ( and ). Taken together, these results indicated that BECN2 targeted inflammasome sensors for degradation through a lysosomal pathway.
BECN2 degrades inflammasome sensors through ULK1/ATG9A-dependent but ATG16L1/LC3/BECN1/WIPI2-independent non-canonical autophagy
It has been known that classical autophagy degradation pathway is controlled by a set of evolutionarily conserved ATG (autophagy related) proteins in a process that includes: (i) initiation of the phagophore formation through MTOR (mechanistic target of rapamycin kinase); (ii) nucleation through class III phosphatidylinositol 3-kinase complex (PtdIns3K complex); (iii) expansion to form the autophagosomes through ATG12-, ATG7-, ATG5-, ATG16L1- and LC3/Atg8-controlled ubiquitin-like conjugation systems; and (iv) maturation and degradation via fusion with lysosomes [Citation3,Citation27]. To further determine whether BECN2 mediated the degradation of inflammasome sensors through autophagy, we knocked out a series of key proteins involved in the classical autophagy pathway. We unexpectedly found that BECN2-mediated degradation of AIM2 and NLRP3 was not compromised or impaired in the cells deficient in BECN1, ATG14, PIK3R4/VPS15, ATG7, ATG16L1, WIPI2, or LC3B (, and S3A-F). By contrast, the ablation of ULK1 and ATG9A could markedly inhibit the BECN2-mediated degradation of AIM2 and NLRP3 (). Consistently, BECN2-mediated degradation could be blocked by the ULK1 complex inhibitors SBI-0206965 and MRT68921 (Fig. S4A). These results indicated that BECN2-mediated degradation of inflammasome sensor proteins required a ULK1- and ATG9A-dependent, but BECN1, ATG14, PIK3R4, ATG7, ATG16L1, WIPI2, and LC3B-independent non-canonical autophagic degradation pathway. Since the inflammasome sensors can also be degraded by canonical autophagy [Citation1–5], we first examined the modulation of canonical autophagy by BECN2, and found that both LC3B turnover and SQSTM1/p62 degradation were only slightly affected by BECN2 deficiency, compared to ATG16L1 deficiency (Fig. S4B). Consistently, overexpression of BECN2 had little effect on the LC3B turnover and SQSTM1 degradation, suggesting a minor role of BECN2 in the regulation of canonical autophagy (Fig. S4C). We next compared the protein levels of NLRP3 and AIM2 in WT, BECN2 KO, and ATG16L1 KO THP1 cells, and found that the protein levels of NLRP3 and AIM2 in BECN2 KO cells were increased to a level similar to those in ATG16L1 KO cells based on the quantification of the blots, suggesting that NLRP3 and AIM2 were degraded through BECN2-mediated non-canonical autophagy and ATG16L1-mediated canonical autophagy at the similar level (Fig. S4D). We further compared the loss of BECN2 to the deficiency of other classical autophagy components, including BECN1 and ATG16L1, in inflammasome hyperactivation, and found that IL1B levels were significantly enhanced in all KO cells, compared to WT cells (Fig. S4E). Notably, similar amounts of IL1B were produced from BECN2 KO and ATG16L1 KO THP1 cells. Importantly, both BECN2 KO and ATG16L1 KO cells produced significantly higher amounts of IL1B than BECN1 KO cells (Fig. S4E).
Figure 4. BECN2-mediated degradation of inflammasome sensors is through the ULK1/ATG9A-dependent but BECN1/ATG16L1/ATG7/LC3B-independent lysosomal pathway. (A-J) WT and autophagy gene KO HEK293T cells were co-transfected with HA-tagged BECN2 and Flag-tagged NLRP3 or AIM2. Protein levels of AIM2 and NLRP3 were immunoblotted using indicated antibodies in BECN1 KO cells (A and B), ATG16L1 and ATG7 KO cells (C and D), MAP1LC3B KO cells (E and F), ULK1 KO cells (G and H), and ATG9A KO cells (I and J). Quantification analysis is presented as mean ± s.e.m. and is calculated based on the band density of three independent experiments. Statistical differences between EmpVec-transfected and HA-BECN2-tranfected cells were calculated using Student’s unpaired t-test (A-J). Statistical differences between WT and KO groups were calculated using Student’s unpaired t-test (A, B, E, F) or 1-way ANOVA with Dunnett’s multiple comparison test (C, D, G-J). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
ATG9A functions as a central dock to bring inflammasome sensor proteins and BECN2 together
To determine the role of ULK1 and ATG9A in BECN2-mediated degradation of inflammasome sensors, we performed co-immunoprecipitation experiments and found that BECN2 could interact with ATG9A and ULK1, but not other components in ULK1 complex (including RB1CC1/FIP200 and ATG13) (Fig. S5A), which is consistent with our recent report that demonstrates the endogenous interactions of BECN2 with ULK1 and ATG9A [Citation13]. We sought to determine which domain of BECN2 was required for its interaction with ATG9A, and found that the BECN2 CCD-ECD domain, which was the same domain required for interacting with AIM2 and NLRP3, was responsible for its interaction with ATG9A (Fig. S5B). We next examined whether the interactions between BECN2 and inflammasome sensors could be abolished in the ULK1- or ATG9A-deficient cells. Indeed, we found that interactions between BECN2 and AIM2 or NLRP3 were markedly reduced in ULK1- and ATG9A-deficient cells, compared to WT cells ( and ). The confocal microscopic analyses further revealed that mCherry-BECN2 failed to be co-localized with GFP-AIM2 in ULK1 or ATG9A KO HEK293T cells (). Consistently, the transportation of AIM2 to lysosome was largely impaired in ULK1 KO and ATG9A KO cells, compared to WT cells (Fig. S5C).
Figure 5. The interaction between BECN2 and inflammasome components requires ULK1 and ATG9A. (A and B) WT, ULK1 KO, and ATG9A KO HEK293T were co-transfected with HA-tagged BECN2 and Flag-tagged AIM2 (A) or NLRP3 (B), the cell lysates were immunoprecipitated with anti-Flag beads and then immunoblotted using indicated antibodies. (C) WT, ULK1 KO and ATG9A KO HEK293T cells co-transfected with GFP-AIM2 and mCherry-BECN2 were fixed with 4% (wt/vol) paraformaldehyde. Pearson’s correlation coefficient for colocalization was analyzed using Image J Coloc 2. Graph is plotted as mean ± s.e.m. (at least 30 cells were analyzed per condition). Representative images showing the colocalization were obtained using confocal microscopy. Scale bar: 5 μm. (D and E) WT, BECN2 KO, and ULK1 KO HEK293T were co-transfected with HA-tagged ATG9A and Flag-tagged AIM2 (D) or NLRP3 (E), the cell lysates were immunoprecipitated with anti-Flag beads and then immunoblotted using indicated antibodies. (F) WT and Becn2 KO BMDMs cells were left untreated or stimulated with LPS (200 ng/ml) for 3 h, the cell lysates were then immunoprecipitated using ATG9A antibodies, followed by immunoblotting with indicated antibodies. (G) WT and ULK1 KO HEK293T cells co-transfected with GFP-AIM2 and mCherry-ATG9A were fixed with 4% (wt/vol) paraformaldehyde. Pearson’s correlation coefficient for colocalization was analyzed using Image J Coloc 2. Graph represents mean ± s.e.m. (at least 30 cells were analyzed per condition). Representative images showing the colocalization were obtained using confocal microscopy. Scale bar: 5 μm. (H) WT and ULK1 KO HEK293T were co-transfected with Flag-tagged BECN2 and HA-tagged ATG9A, the cell lysates were immunoprecipitated with anti-Flag beads and then immunoblotted using indicated antibodies. Data are representative of at least three independent experiments. (I) WT and BECN2 KO 293T cells were transfected with AIM2-APEX2 encoding plasmid alone or along with Flag-BECN2. Representative EM images of cells expressing AIM2-APEX2 were processed in parallel under identical conditions. Black arrow indicates the AIM2 location. AP, autophagosome or amphisome; AL, autolysosome. Statistical differences between WT and KO groups were calculated using Student’s unpaired t-test (G) or 1-way ANOVA with Dunnett’s multiple comparison test (C). ***P < 0.001.
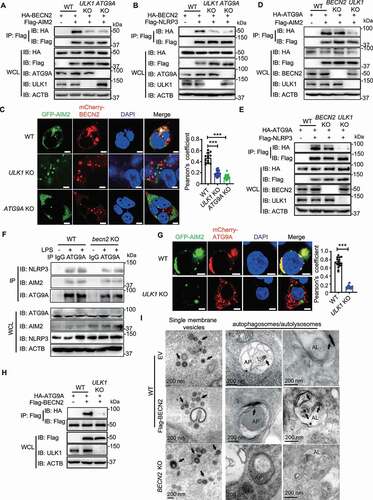
To further understand how ULK1 and ATG9A control the interaction of BECN2 and its target proteins (inflammasome sensors), we performed co-immunoprecipitation experiments and found that ATG9A could directly interact with AIM2 or NLRP3 in WT and BECN2-deficient cells, but not in ULK1-deficient cells ( and ). Consistently, the endogenous interactions of ATG9A with NLRP3 and AIM2 remained intact in BMDMs derived from WT and Becn2-deficient mice regardless of LPS stimulation (). Confocal microscopic studies demonstrated that the colocalization between AIM2 and ATG9A was significantly compromised in ULK1 KO cells (). Likewise, the interaction between BECN2 and ATG9A was also ULK1-dependent (). These results suggest that ULK1 is essentially required for ATG9A to serve as a central dock to interact with target proteins (inflammasome sensors) and BECN2. However, our further experiments showed that ULK1 and its associated proteins (RB1CC1 and ATG13) did not interact with AIM2 or NLRP3 (Fig. S5D and S5E). These results suggest that the ULK1 complex is required for activating ATG9A, which, in turn, interacts with and bring target proteins and BECN2 together, but is not required for the direct interaction with inflammasome sensor proteins.
To further understand the process of ATG9A-BECN2-mediated inflammasome sensor degradation, we next examined the translocation of AIM2 by transmission electron microscopy (TEM) in WT, BECN2-overexpressed WT cells, and BECN2 KO 293 T cells through APEX2-enabled staining. We found that AIM2 was frequently associated with single-membrane vesicles and autophagosomes or amphisomes (intermediate organelles formed by fusion of autophagosomes and endosomes) in WT cells (). The presence of APEX2-AIM2 in autophagosomes/amphisomes was significantly increased after BECN2 overexpression. However, AIM2 was barely detected in autophagosomes in BECN2 KO cells, but mainly associated with single-membrane vesicles (). These observations suggest that BECN2 could facilitate the AIM2-associated vesicles fusion with phagophore assembly site or autophagosomes for autophagic degradation.
BECN2-mediated degradation of inflammasome sensors requires SEC22A-STX5-STX6 SNAREs
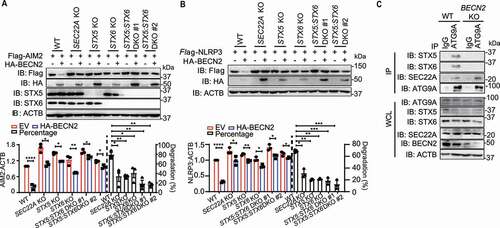
ATG9A is a multispanning transmembrane ATG protein that cycles between the trans-Golgi network (TGN) and endosomes/lysosomes. The ATG9A+-vesicle recruitment to phagophore assembly site is also an important resource for the membrane assembling during autophagosome formation [Citation28–31]. Based on our data showing that AIM2 was frequently associated with single membrane vesicles (presumably to be ATG9A-vesicles) and could be markedly transported to autophagosomes after BECN2 overexpression, we reasoned that BECN2 might promote the degradation of inflammasome sensors through multiple steps of vesicle membrane fusions. RABs and soluble NSF attachment protein receptors (SNAREs) machinery have been reported to play a central role in membrane docking/fusion process [Citation32–34]. To test this possibility, we examined the potential interactions between BECN2 and some members of SNAREs and RAB GTPases. Co-immunoprecipitation experiments showed that BECN2 could strongly interact with RAB8A, SEC22A, STX5 (syntaxin 5), STX6, STX7, STX8, VAMP7, and VAMP8, and weakly interact with RAB7A, RAB32, VTI1B, and STX17 (Fig. S6A). We next investigated whether the deficiency of these BECN2-binding proteins could completely or partially block the inflammasome sensor degradation. For this reason, we generated STX5, STX6, or SEC22A KO cell lines, and found that specific ablation of STX5, STX6, or SEC22A in HEK293T cells using CRISPR-Cas9 technology could partially block the BECN2-mediated AIM2 or NLRP3 degradation (, , and S6B). By contrast, KO of other RAB or SNARE genes had little or no effect on NLRP3 degradation (Fig. S6C and 6D). To further determine their potential effects, we generated STX5 and STX6 double KO (DKO) cells and showed that the DKO of STX5 and STX6 had much stronger inhibitory effects on BECN2-mediated inflammasome sensor degradation than single KO ( and ). To determine the role of BECN2 in STX5-STX6-SEC22A-mediated ATG9A-vesicle membrane fusion, we also examined the endogenous interaction between ATG9A and STX5-STX6-SEC22A in WT and BECN2 KO THP-1 cells. The endogenous interactions of ATG9A with STX5 and STX6 were detected in WT THP-1 cells, but diminished by the ablation of BECN2. By contrast, the interaction between ATG9A and SEC22A remained unchanged regardless of BECN2 expression (). These data suggest that BECN2 is required for the interaction of ATG9A vesicles with STX5-STX6 SNAREs for the degradation of inflammasome sensor proteins through vesicle membrane fusion processes (Fig. S6E).
Figure 6. SEC22A, STX5, and STX6 are required for BECN2-mediated degradation of inflammasome components. (A and B) WT, SEC22A KO, STX5 KO, STX6 KO, and STX5:STX6 DKO HEK293T cells were co-transfected with HA-tagged BECN2 and Flag-tagged AIM2 (A) or NLRP3 (B). Protein levels of AIM2 and NLRP3 were immunoblotted using indicated antibodies. Quantification analysis is presented as mean ± s.e.m. and is calculated based on the band density of three independent experiments. (C) WT and BECN2 KO THP-1 cells were lysed and immunoprecipitated using ATG9A antibodies, followed by immunoblotting with indicated antibodies. Statistical differences between EmpVec-transfected and HA-BECN2-transfected cells were calculated using Student’s unpaired t-test (A and B). Statistical differences of degradation percentages between WT and KO groups were calculated using 1-way ANOVA with Dunnett’s multiple comparison test (A and B). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Data are mean ± s.e.m. of three independent experiments.
Loss of BECN2 exacerbates alum-induced peritonitis
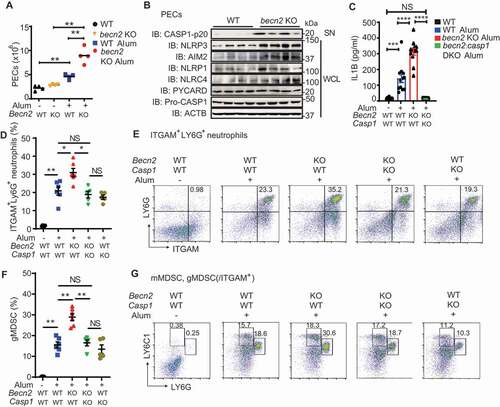
To further substantiate these findings in vivo, we investigated the phenotypes of becn2 KO mice in alum-induced peritonitis model [Citation35,Citation36]. Mouse peritonitis was induced by an intraperitoneal injection of alum at a dose of 700 μg/mouse. We collected peritoneal exudate cells (PECs) by peritoneal lavage and found a significant increase in the total number of PECs in becn2 KO mice (). The protein levels of NLRP3, AIM2, NLRP1, NLRC4, as well as the cleavage of CASP1 were all dramatically increased in PECs from becn2 KO mice, as compared with WT controls (). Nevertheless, no appreciable difference was observed in the protein levels of pro-CASP1 and PYCARD (). Consistently, the IL1B levels were significantly increased in the lavage fluid from becn2 KO mice after alum challenge (). Furthermore, the neutrophils (ITGAM/CD11b+LY6G+) and granulocytic myeloid-derived suppressor cells (gMDSC, ITGAM+LY6C1intLY6Ghigh) subsets were both significantly elevated in the peritoneal fluid from alum-treated Becn2-deficient mice, whereas the percentage of monocytic MDSC (mMDSC, ITGAM+LY6C1highLY6Glow) did not show an appreciable difference among different genotypes (). To further determine whether the deletion of Casp1, the downstream effector for all inflammasomes, could rescue the enhanced peritonitis in alum-challenged Becn2-deficient mice, we crossed casp1 KO mice with becn2 KO mice to generate becn2:casp1 DKO strain. The successful deletion of casp1 was validated by PCR analysis (Fig. S6F). We showed that the increased IL1B production and percentages of neutrophils and gMDSC in peritoneal lavage fluid from Becn2-deficient mice were restored in DKO mice to the levels similar to those in WT controls (). Taken together, these data suggested that loss of BECN2 exacerbated alum-induced peritonitis by promoting the inflammasome activation and the recruitment of excessive inflammatory immune cells.
Figure 7. Loss of BECN2 exacerbates alum-induced peritoneal inflammation. (A, B) WT and becn2 KO mice were i.p. injected with alum (700 μg). The peritoneal exudate cells (PECs) were collected 12 h after injection. (A) Absolute numbers of PECs recruited to the peritoneum were counted (n = 4). (B) Protein levels of pro-CASP1 and inflammasome sensors in whole cell lysates (WCL) and cleaved CASP1 in SN of PECs at 12 h post-ex vivo culture was detected by immunoblot using indicated antibodies. (C-G) WT, becn2 KO and becn2: casp1 DKO mice were i.p. injected with alum (700 μg). IL1B production in peritoneal lavage obtained at 12 h post-injection was determined by ELISA (C). Data are mean ± s.e.m. of three independent experiments. Percentage of ITGAM+ LY6G+ neutrophils (D and E), monocytic (mMDSC, ITGAM+ LY6C1high LY6Glow) and granulocytic (gMDSC, ITGAM+ LY6C1int LY6Ghigh) subsets of myeloid-derived suppressor cells (F and G) recruited to the peritoneum were analyzed by flow cytometry. Statistical differences between groups were calculated using 1-way ANOVA with Tukey’s multiple comparison test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Discussion
In this study, we identified the critical role of BECN2 in the negative regulation of inflammasome activation. Genetic deletion of BECN2 enhanced the activities of inflammasomes in response to NLRP3, AIM2, NLRC4, and NLRP1 ligand stimulations, while overexpression of BECN2 suppressed the inflammasome activation. Our recent study demonstrated that BECN2 functioned as a negative regulator in the control of the MAPK1/ERK2-MAPK3/ERK1 and NFKB signaling pathways [Citation13]. Since IL1B is generated from the proteolytic cleavage of its precursor protein pro-IL1B by active CASP1, the enhanced IL1B production from Becn2 KO cells and becn2 KO mice are assumed to be contributed by both increased NFKB signaling for pro-IL1B expression and enhanced inflammasome activity for generating mature IL1B. Previous reports suggest that autophagy protein functions in the removal of inflammasome components through selective autophagy by SQSTM1 recognition of K63 (Lys 63)-linked polyubiquitinated PYCARD [Citation1]. Additionally, BECN1, a homolog of BECN2, is involved in the removal of damaged mitochondria through autophagy, and the loss of BECN1 enhances the inflammasome activity due to excessive mtDNA [Citation32,Citation37]. We compared the loss of BECN2 to the deficiency of BECN1 and ATG16L1, and found that IL1B productions were significant enhanced in BECN2 KO to a level similar to ATG16L1 KO THP1 cells. Consistently, the protein levels of NLRP3 and AIM2 in BECN2 KO cells were increased to a level similar to those in ATG16L1 KO, suggesting an indispensable role of BECN2-mediated non-canonical autophagy in inflammasome regulation compared to classical autophagy. The degradation of inflammasome sensors by BECN2 was demonstrated both in vitro and in vivo, as the protein levels of inflammasome sensors were markedly higher in macrophages isolated becn2 KO mice compared with cells from WT mice. Importantly, we demonstrated that BECN2 targets inflammasome sensors for degradation through an ATG9-dependent, but ATG16L1/LC3/BECN1-independent autophagic pathway.
Using cell lines with knockout of key ATG genes by CRISPR/Cas9 technology, we showed that BECN2-mediated degradation of inflammasome sensors was independent of BECN1/ATG14/PIK3R4-mediated nucleation or ATG16L1/ATG7/LC3-controlled ubiquitin-like conjugation systems, but rather relied on ULK1 and ATG9A. BECN2 interacted with inflammasome sensors through its CCD-ECD domain, but such interactions could be remarkedly disrupted in ATG9A- and ULK1-deficient cells. Because ULK1 is a serine/threonine kinase and stimulates autophagy via the phosphorylation of ATG9A [Citation30,Citation31], we reasoned that the phosphorylated ATG9A might serve as a platform for bridging BECN2 and inflammasome sensors. Consistently, we showed that neither inflammasome sensors nor BECN2 could interact with ATG9A in ULK1 KO cells, indicating the requirement of ULK1 for initiating the assembly of inflammasome sensors-ATG9A-BECN2 complex. Furthermore, inflammasome sensors and ATG9A remained associated in BECN2 KO cells, suggesting that their interaction is independent of BECN2. Thus, ATG9A functioned as a central dock to bring BECN2 with AIM2 or NLRP3 together.
ATG9A is the only transmembrane ATG protein that mainly localizes to the trans-Golgi network (TGN) and endosomes, and serves as an important membrane resource for autophagosome initiation [Citation28–31,Citation38]. It has been demonstrated that ULK1 is critically important to activate ATG9 through multiple phosphorylation sites in yeast and mammalian cells, thus regulating its trafficking and role in the early phase of autophagosome formation [Citation28,Citation31,Citation38–41]. However, the role of ATG9 in non-canonical autophagic degradation pathway has not yet been well defined. We demonstrated that ATG9A-BECN2-dependent inflammasome sensor degradation was independent of BECN1/ATG16L1/ATG7/LC3-mediated classical autophagic pathway. Non-canonical autophagy has been demonstrated to involve in the regulation of type I interferon signaling and B cell responses upon viral infection [Citation27,Citation42,Citation43]. The non-canonical autophagy could bypass some steps in canonical autophagy through the recruitment of isolated membranes from multiple resources [Citation27,Citation42,Citation43]. Unlike the autophagy-independent lysosome degradation of G protein-coupled receptors by BECN2 [Citation11,Citation12], our current study showed that BECN2 played a critical role in a non-canonical autophagic degradation of inflammasome sensors [Citation13]. This notion is supported by our recent study showing that BECN2 targets MAP3K7/TAK1 and MAP3K3/MEKK3 for degradation through an ATG9-dependent, but BECN1/ATG16L1/LC3 independent, non-canonical autophagic pathway [Citation13]. Although ATG9A is known to be embedded in the autophagosomal outer membrane after fusion with phagophores [Citation31], our TEM study clearly showed that inflammasome sensors were delivered into the phagophore/autophagosome lumen upon the completion of the fusion. Furthermore, AIM2 could be frequently delivered to autophagosomes after BECN2 overexpression, while such translocation was impeded by the ablation of BECN2. These observations suggest that BECN2 might facilitate the autophagic degradation of AIM2 through the fusion of AIM2-associated ATG9A+-endosomes with phagophores/autophagosomes. Since inflammasome sensors might be disassociated with ATG9A probably due to the conformation changes during the fusion process, AIM2 was retained within the phagophores/autophagosomes (Fig. S6F). TEM results further showed that the inflammasome sensor-containing autophagosomes could fuse with lysosomes and form autolysosomes for degradation. In addition, emerging roles of RAB and SNARE proteins have been recently shown in the membrane fusion process during autophagy, such as STX17 in the fusion of autophagosomes with lysosomes in cells lacking ATG-conjugation system [Citation34]. SNARE family proteins mainly function to drive membrane fusion, in which one part of the SNAREs embeds in the vesicle membrane (v-SNAREs) and the other part in the target site (t-SNAREs) [Citation32,Citation37]. Matched pairs of v- and t-SNAREs interact and pull the opposing membranes into a closer association for fusion [Citation32,Citation37]. Previous reports show that STX5 and STX6 function as t-SNAREs to drive the fusion of autophagosomes and lysosomes, as well as the fusion between endosomes and autophagosomes, respectively [Citation44,Citation45]. We recently identified STX5 and STX6 as essential SNAREs that are required for BECN2-mediated non-canonical autophagic degradation of MAP3K7 and MAP3K3 [Citation13]. DKO of STX5 and STX6 blocks the transportation of ATG9A+-associated MAP3K3 to autophagosomes for degradation [Citation13]. Consistent with our previous finding, herein we showed that STX5, STX6 and SEC22A were critically required for BECN2-mediated degradation of inflammasome sensors. We demonstrated that the interaction between ATG9A and STX5-STX6 were BECN2-dependent, suggesting the critical role of BECN2 in mediating the STX5- and STX6-dependent degradation of inflammasome sensors through membrane fusion. These data collectively implied that the BECN2-mediated non-canonical autophagic pathway might be engaged in degrading a variety of proteins for the maintenance of their cellular hemostasis. However, the detailed mechanisms by which STX5, STX6 and SEC22A are involved in the membrane fusion of BECN2+ATG9A+-vesicles with phagophores or autophagosomes for inflammasome sensor degradation warrant further investigation.
Inflammasome activation must be tightly regulated, otherwise, uncontrolled inflammasome activation could lead to autoinflammatory syndromes, metabolic diseases, and neurodegenerative diseases [Citation15,Citation18]. Our findings illustrated the in vivo physiological relevance of BECN2 in regulating inflammasome-mediated inflammation via alum-induced peritonitis model. We observed the elevated IL1B in peritoneal lavage fluid, the increased inflammatory neutrophils and granulocytic myeloid-derived suppressor cells (gMDSC, ITGAM+LY6C1intLY6Ghigh), as well as enhanced cleavage of CASP1 in becn2 KO PECs, compared with WT controls. We recently found that the IL1B precursor (pro-IL1B) levels were increased in becn2 KO cells, compared to that in WT cells, after LPS treatment, owing to the elevated NFKB signaling [Citation13]. Therefore, both NFKB signaling and inflammasome pathways might contribute to the markedly enhanced IL1B production in becn2 KO immune cells and in Becn2-deficient mice. We further demonstrated that the ablation of Casp1 in becn2 KO mice could restore the alum-induced peritonitis to a level similar to WT mice. Overall, our study has identified a previously unrecognized role of BECN2 in the negative regulation of inflammasome activation by targeting inflammasome sensors for degradation. Mechanistically, BECN2 interacted with inflammasome sensors to direct them to lysosomes for degradation through ULK1- and ATG9A-dependent non-classical autophagy. In particular, we showed that SEC22A, STX5, and STX6 played essential roles in BECN2-mediated inflammasome sensor degradation. Therefore, our findings have identified a previously unrecognized role of BECN2 in the negative regulation of several key inflammasome sensor proteins, and provided molecular insights into the potential mechanisms by which BECN2 mediates inflammasome sensors for degradation in a ULK1- and ATG9A-dependent non-canonical autophagic pathway. Our finding suggests that BECN2 and its interacting proteins may serve as potential therapeutic targets for the prevention and treatment of inflammation-associated diseases.
Materials and methods
Antibodies and reagents
Antibodies and reagents used in this study were purchased from different suppliers as indicated in Key resources table ().
Table 1. Key resources table
Mouse experiment
All animal experiments were performed in animal housing facilities under specific pathogen-free conditions at Houston Methodist Research Institute. All animal studies were performed according to the NIH guidelines for the use and care of live animals and approved by the Animal Care and Use Committee of the Houston Methodist Research Institute. Wild-type and casp1 KO C57BL/6 mice were obtained from Jackson Laboratory, becn2 KO mouse was kindly provided by Dr. Beth Levine (University of Texas Southwestern Medical Center, Dallas; deceased). We maintained both becn2 and casp1 heterozygous and homozygous KO mice for breeding. becn2 KO mice were generated from both becn2 heterozygous and homozygous KO breeding pairs, and becn2 casp1 DKO mice were generated from becn2 heterozygous casp1 homozygous KO breeding pairs. All mice were 6–8 weeks of age for experimental use. For the in vivo LPS treatment, mice were i.p. injected with LPS (30 mg/kg body weight; Sigma-Aldrich, L4391) and blood samples were collected to examine the serum IL1B levels. For induction of peritonitis, WT, Becn2-deficient, and Casp1-deficient mice (females, 6-weeks old) were i.p. injected with 700 μg alum/mouse. To analyze the IL1B levels and PECs in the peritoneal cavity, peritoneal cavities were lavaged with cold PBS (Thermo Fisher Scientific, 10,010,023) at 12 h post i.p. injection of alum.
Cell culture
HEK293T (CRL-3216), THP-1 cells (TIB-202) and HeLa cells (CRM-CCL-2) were purchased from American Type Culture Collection (ATCC). HEK293T and HeLa cells were grown in DMEM (Gibco, 11,965–092) supplemented with 10% fetal bovine serum and 0.5% penicillin-streptomycin (Sigma-Aldrich, V900929). THP-1 cells were cultured in RPMI-1640 medium (Gibco, 11,875–093) supplemented with 10% fetal bovine serum (Sigma-Aldrich, F8318), 0.5% penicillin-streptomycin, and 0.05 mM 2-mercaptoethanol (Sigma-Aldrich, M6250). Mouse bone marrow cells were isolated from mouse femurs and tibias and differentiated into bone marrow-derived macrophages (BMDMs) with L929 cell-conditioned medium (ATCC, CRL-6364) for 5–6 d. For inflammasome activation studies, BMDMs, HEK293T-CIA and THP-1 cells were primed for 3 h with LPS (100 ng/ml), then stimulated with nigericin (Invivogen, tlrl-nig; 1 μM, 6 h), poly(dA:dT) (Invivogen, poly[(dA:dT)]/LyoVec, tlrl-patn; 1 μg/ml, 6 h), anthrax lethal factor (LF) (List Lab, recombinant from B. anthracis; 172D; 1 μg/ml, 4–6 h), or flagellin (Invivogen, FLA-ST; tlrl-stfla; 20 μg/ml, 6–8 h). For cycloheximide chase assays, cells were treated with cycloheximide (Cell Signaling Technology, 2112) at 100 mg/ml for different time points starting from 24 h post-transfection.
Cloning and plasmids
Plasmids encoding human BECN2, AIM2, NLRP3, NLRC4, NLRP1, PYCARD, CASP1, ATG9A, RAB4A, RAB7A, RAB8A, RAB9A, RAB11A, RAB24, RAB32, RAB33B, SNAP25, SNAP29, SEC22B, SEC22A, STX5, STX6, STX7, STX8, STX17, VAMP4, VAMP7, VAMP8, and VTI1B were generated from Human ORFeome library and/or Baylor Ultimate ORF LITE using the gateway cloning system (Thermo Fisher Scientific, S7020), or cloned using HEK293T cDNA (see primer sequence in ). Vectors including pcDNA-ccdB-FLAG, pcDNA-ccdB-HA, pcDNA-ccdB-eGFP or pcDNA-mCherry2 vectors were transcribed under the control of the CMV promoter. Truncation of NLRP3 (PYD 1–94, NACHT 95–559, LRR 560–894), AIM2 (PYD 1–116, HIN 117–343), BECN2 (N 1–87, ΔN 88–431, CCDECD 110–431, ΔECD 1–248) were generated by PCR using primers listed in and subcloned into final constructs containing affinity tag. The plasmid encoding pUC57-APEX2 was obtained from Addgene (40,306; deposited by Alice Ting). AIM2-APEX2 encoding sequence was cloned into pcDNA-3.1 vectors by homologous recombination using NEBuilder® HiFi DNA Assembly Cloning Kit (New England BioLabs, E5520S).
Table 2. Sequences of primers used for constructing gene expression plasmids, related to Methods
Immunoblot analyses
Cells were lysed using RIPA buffer (150 mM NaCl, 1.0% IGEPAL CA-630 [Sigma-Aldrich, I8896], 0.5% sodium deoxycholate [Sigma-Aldrich, 30,970], 0.1% SDS, 50 mM pH 8.0 Tris-HCl, protease inhibitor cocktail [Roche, 4,693,159,001]) on ice for 15 min. Immunoblotting was performed by loading the samples on SDS-PAGE gels, conducting electrophoresis, transferring the samples to PVDF membranes (Bio-Rad, 1,620,177), and then incubating the membranes with the indicated antibodies. For all immunoblots, the Luminata western HRP Chemiluminescence Substrates (Millipore, WBKLS0500) and ChemiDoc XRS+ System with Image Lab (Bio-Rad) was used for protein detection.
Flow cytometry
Mouse peritoneal exudates cells (PECs) were obtained from peritoneal lavage fluid and stained with indicated antibodies for cell surface staining for 20 min at 4°C in PBS containing 1% FCS and 10 mM EDTA. Anti-ITGAM, anti-LY6G, and anti-LY6C1 (Thermo Fisher Scientific, 12–0112-82, 11–9668-82 and 17–5932-82) were used to label neutrophils. Monocytic myeloid-derived suppressor cells (mMDSC) were characterized as ITGAM+LY6C1highLY6Glow, and granulocytic MDSC (gMDSC) was characterized as ITGAM+LY6C1intLY6Ghigh. Flow cytometric analysis was performed with a BD FACSCalibur or BD FACSAria system (Becton Dickson). The acquired data were analyzed with FlowJo software.
Immunoprecipitation and supernatant protein precipitation
For IP experiments, whole cell lysates obtained 24 h post-transfection or via ligand stimulation were obtained using RIPA lysis buffer (as described previously) and shaken on ice for 15 min. The HEK293T cell lysates were immunoprecipitated with anti-Flag conjugated beads (Sigma-Aldrich, M8823) at 4°C overnight. The lysates of THP-1 cells or BMDMs were immunoprecipitated with 2 µg/ml anti-AIM2, anti-NLRP3 antibody, or anti-ATG9A antibody along with protein A/G-agarose beads (Thermo Fisher Scientific, 20,421) at 4°C overnight. The beads were washed five times with RIPA lysis buffer, and immunoprecipitates were eluted with 2X SDS loading buffer and subjected to SDS-PAGE gel electrophoresis, followed by immunoblotting. The secondary antibody (Veriblot for IP detection, HRP-conjugated) that only recognized native IgG but not denatured IgG was applied for immunoblotting of endogenous proteins in immunoprecipitates. For immunoblotting of the cleaved CASP1 in cell culture supernatants, supernatants (400 µl) were precipitated by add 400 µl methanol and 100 µl chloroform, then vortexed and centrifuged for 15 min at 14,000 g. The upper phase was discarded and another 400 µl methanol was added to the interphase. This mixture was centrifuged for 15 min at 14,000 g and the protein pellet was dried at room temperature, resuspended with 2X SDS loading buffer and boiled for 5 min at 95°C.
ELISA for cytokine production
Bone marrow macrophages, HEK293T-CIA cells or THP-1 cells were seeded in 24-well plates and cultured overnight. After priming with 200 ng/ml LPS for 3 h and stimulating with nigericin (1 μM, 6 h), poly(dA:dT) (1 μg/ml, 6 h), anthrax lethal factor (LF) (1 μg/ml, 4–6 h) or flagellin (20 μg/ml, 6–8 h), the supernatants were collected and measured for IL1B concentrations using IL1B ELISA kits (human interferon beta ELISA kit, PBL, eBioscience, 41,410; or anti-mouse IL1B ELISA kit, eBioscience, E05277-1531; 1:500 dilution, E03232-1632; 1:1000 dilution) according to the manufacturer’s protocols.
Gene knockout by CRISPR/Cas9 technology
We designed human BECN2, BECN1, ATG16L1, ATG7, MAP1LC3B, ATG14, WIPI2, PIK3R4/VPS15, STX5, STX6, SEC22A, ULK1, and ATG9A sgRNAs using an online CRISPR design tool (crispr.mit.edu) by inputting targeted exon sequence. VAMP7, VAMP8, RAB7A, RAB8A, RAB32, STX7, STX8, STX17, VT1LB sgRNAs were from Library (LentiArray human CRISPR library, Thermo Fisher Scientific). Designed sgRNAs were cloned into the BsmB1 site of pLenti-Crispr-Cas9 v2 vectors (Addgene, 52,961, deposited by Feng Zhang) containing Cas9-P2Apuromycin and were verified by sequencing analysis. The sgRNA-containing plasmids were transfected into HEK293T cells with psPAX2 (Addgene, 12,260, deposited by Didier Trono) and pMD2.G (Addgene, 12,259, deposited by Didier Trono) plasmids. After two days, the virus-containing medium was subjected to ultracentrifugation (20,000 g at 4°C for 2 h) and frozen at −80°C. HEK293T cells were transduced with control sgRNA- or gene-targeting sgRNA-containing lentiCRISPR viruses. Transduced cells were selected in the presence of puromycin (Invivogen, ant-pr-1) at 2 µg/ml for 3 d. The KO efficiency was confirmed by immunoblot analysis or genomic DNA sequencing. A list of sgRNA sequences for KO is presented in .
Table 3. Design of the single-guide RNA sequence (sgRNA) for gene knockout by the Cas9-expression vector pLentiCRISPR-v2, related to Methods
Immunofluorescence staining
HeLa or HEK293T cells were fixed for 15 min with 4% paraformaldehyde 24 h post-transfection and then permeabilized in methyl alcohol for 10 min at −20°C. After washing three times with PBS, fixed cells were blocked in 10% normal goat serum for 1 h, incubated with primary antibody overnight, and incubated with goat anti-mouse IgG, Alexa Fluor 594 secondary antibodies (Thermo Fisher Scientific, A11032). Nuclei were stained with DAPI (Abcam, ab104139; 1:1,000 dilution) or Hoechst 33,342 (Abcam, ab228551; 1:1,000 dilution) at 24 h post-transfection. Lysosome was stained with LysoTracker-blue (Life Technology, 1,662,594; 1:1,000 dilution, 40 min, 37°C) 24 h post-transfection, then washed twice with fresh medium. Microscopy was performed using a confocal microscope (Olympus, FV3000).
Transmission electron microscopy (TEM) and APEX2-enabled AIM2 staining
For EM imaging of AIM2-APEX2, WT and BECN2 KO 293 T cells were transfected with AIM2-APEX2-encoding plasmid alone or along with Flag-BECN2. At 24 h post-transfection, cells were fixed in 2.5% (vol:vol) glutaraldehyde and processed for staining. Briefly, cells were fixed with Karnovsky’s fixative (Fisher Scientific, 50–980-496), incubated in 3,3ʹ-diaminobenzidine (DAB; Sigma-Aldrich, 91–95-2) solution, followed by post-fixation in osmium tetroxide and stained with uranyl acetate. After dehydrated in a series of graded ethanol and embedded in epoxy resin, samples were sectioned at 100-nm thickness using a Leica EM UC7 ultramicrotome. Electron micrographs were obtained under a JEOL JEM-1230 TEM equipped with a Gatan CCD camera.
RNA extraction and real-time PCR
Total RNA was extracted from cells or homogenized tissues using Trizol reagent (Invitrogen, 15,596,026) according to the manufacturer’s protocol. For RT-PCR analysis, cDNA was generated using SuperScript III reverse transcriptase (Thermo Fisher Scientific, 18,080,093) and analyzed through quantitative RT-PCR using the SYBR green PCR master mix (Thermo Fisher Scientific, 4,367,659) on a QuantStudio 6 flex real-time PCR system (Applied Biosystems). All the data were normalized to GAPDH expression. Primer sequences for RT-PCR analysis of gene expression for human AIM2 (F: TCAAGCTGAAATGAGTCCTGC; R: CTTGGGTCTCAAACGTGAAGG), mouse Aim2 (F: GTCACCAGTTCCTCAGTTGTG: R: CACCTCCATTGTCCCTGTTTTAT), human NLRP3 (F: CGTGAGTCCCATTAAGATGGAGT; R: CCCGACAGTGGATATAGAACAGA), and mouse Nlrp3 (F: ATCAACAGGCGAGACCTCTG; R: GTCCTCCTGGCATACCATAGA) are included as above.
Statistical analysis
All the experiments were independently performed at least three times. Results are presented as mean ± s.e.m. as indicated. Statistical analyses were performed using GraphPad Prism v6.0 software with a value of P < 0.05 considered to be statistically significant.
Disclosure of potential conflicts of interest
No potential conflict of interest was reported by the author(s).
Supplemental Material
Download Zip (2.2 MB)Data and materials availability
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Further information and requests for reagents should be directed to the Lead Contact, R.F.W. ([email protected]).
Supplementary material
Supplemental data for this article can be accessed here
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
Related Research Data
References
- Shi CS, Shenderov K, Huang NN, et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13(3):255–263.
- Nakahira K, Haspel JA, Rathinam VA, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12(3):222–230.
- Zhong Z, Sanchez-Lopez E, Karin M. Autophagy, inflammation, and immunity: a troika governing cancer and its treatment. Cell. 2016;166(2):288–298.
- Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456(7219):264–268.
- Sun Q, Fan J, Billiar TR, et al. Inflammasome and autophagy regulation - a two-way street. Mol Med. 2017;23:188–195.
- Heckmann BL, Teubner BJW, Tummers B, et al. LC3-associated endocytosis facilitates beta-amyloid clearance and mitigates neurodegeneration in murine alzheimer’s disease. Cell. 2019;178(3):536–551 e14.
- Galluzzi L, Green DR. Autophagy-independent functions of the autophagy machinery. Cell. 2019;177(7):1682–1699.
- Honda S, Arakawa S, Nishida Y, et al. Ulk1-mediated Atg5-independent macroautophagy mediates elimination of mitochondria from embryonic reticulocytes. Nat Commun. 2014;5:4004.
- Ma T, Li J, Xu Y, et al. Atg5-independent autophagy regulates mitochondrial clearance and is essential for iPSC reprogramming. Nat Cell Biol. 2015;17(11):1379–1387.
- Nishida Y, Arakawa S, Fujitani K, et al. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature. 2009;461(7264):654–658.
- He C, Wei Y, Sun K, et al. Beclin 2 functions in autophagy, degradation of G protein-coupled receptors, and metabolism. Cell. 2013;154(5):1085–1099.
- Dong X, Cheng A, Zou Z, et al. Endolysosomal trafficking of viral G protein-coupled receptor functions in innate immunity and control of viral oncogenesis. Proc Natl Acad Sci USA. 2016;113(11):2994–2999.
- Zhu M, Deng G, Tan P, et al. Beclin 2 negatively regulates innate immune signaling and tumor development. J Clin Invest. 2020. DOI:https://doi.org/10.1172/JCI133283.
- Howrylak JA, Nakahira K. Inflammasomes: key mediators of lung immunity. Annu Rev Physiol. 2017;79:471–494.
- Prochnicki T, Latz E. Inflammasomes on the crossroads of innate immune recognition and metabolic control. Cell Metab. 2017;26(1):71–93.
- Rathinam VA, Fitzgerald KA. Inflammasome complexes: emerging mechanisms and effector functions. Cell. 2016;165(4):792–800.
- Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140(6):821–832.
- Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21(7):677–687.
- Dostert C, Petrilli V, Van Bruggen R, et al. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320(5876):674–677.
- Cassel SL, Eisenbarth SC, Iyer SS, et al. The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci USA. 2008;105(26):9035–9040.
- Hornung V, Bauernfeind F, Halle A, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008 ;9(8):847–856.
- Py BF, Kim MS, Vakifahmetoglu-Norberg H, et al. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell. 2013;49(2):331–338.
- Shenoy AR, Wellington DA, Kumar P, et al. GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science. 2012;336(6080):481–485.
- He Y, Zeng MY, Yang D, et al. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature. 2016;530(7590):354–357.
- Shi H, Wang Y, Li X, et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol. 2016;17(3):250–258.
- Yu JW, Wu J, Zhang Z, et al. Cryopyrin and pyrin activate caspase-1, but not NF-kappaB, via ASC oligomerization. Cell Death Differ. 2006;13(2):236–249.
- Codogno P, Mehrpour M, Proikas-Cezanne T. Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nat Rev Mol Cell Biol. 2011;13(1):7–12.
- Zhou C, Ma K, Gao R, et al. Regulation of mATG9 trafficking by Src- and ULK1-mediated phosphorylation in basal and starvation-induced autophagy. Cell Res. 2017;27(2):184–201.
- Suzuki SW, Yamamoto H, Oikawa Y, et al. Atg13 HORMA domain recruits Atg9 vesicles during autophagosome formation. Proc Natl Acad Sci USA. 2015;112(11):3350–3355.
- Rao Y, Perna MG, Hofmann B, et al. The Atg1-kinase complex tethers Atg9-vesicles to initiate autophagy. Nat Commun. 2016;7:10338.
- Yamamoto H, Kakuta S, Watanabe TM, et al. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J Cell Biol. 2012;198(2):219–233.
- Nair U, Jotwani A, Geng J, et al. SNARE proteins are required for macroautophagy. Cell. 2011;146(2):290–302.
- Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. 2013;14(12):759–774.
- Tsuboyama K, Koyama-Honda I, Sakamaki Y, et al. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science. 2016;354(6315):1036–1041.
- Guarda G, Braun M, Staehli F, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. 2011;34(2):213–223.
- Jin J, Yu Q, Han C, et al. LRRFIP2 negatively regulates NLRP3 inflammasome activation in macrophages by promoting flightless-I-mediated caspase-1 inhibition. Nat Commun. 2013;4:2075.
- Jahn R, Scheller RH. SNAREs–engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7(9):631–643.
- Reggiori F, Shintani T, Nair U, et al. Atg9 cycles between mitochondria and the pre-autophagosomal structure in yeasts. Autophagy. 2005;1(2):101–109.
- Mari M, Griffith J, Rieter E, et al. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J Cell Biol. 2010;190(6):1005–1022.
- Feng Y, Klionsky DJ. Autophagic membrane delivery through ATG9. Cell Res. 2017;27(2):161–162.
- Feng Y, Backues SK, Baba M, et al. Phosphorylation of Atg9 regulates movement to the phagophore assembly site and the rate of autophagosome formation. Autophagy. 2016;12(4):648–658.
- Henault J, Martinez J, Riggs JM, et al. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity. 2012;37(6):986–997.
- Martinez-Martin N, Maldonado P, Gasparrini F, et al. A switch from canonical to noncanonical autophagy shapes B cell responses. Science. 2017;355(6325):641–647.
- Nozawa T, Minowa-Nozawa A, Aikawa C, et al. The STX6-VTI1B-VAMP3 complex facilitates xenophagy by regulating the fusion between recycling endosomes and autophagosomes. Autophagy. 2017;13(1):57–69.
- Renna M, Schaffner C, Winslow AR, et al. Autophagic substrate clearance requires activity of the syntaxin-5 SNARE complex. J Cell Sci. 2011;124:469–482.