
ABSTRACT
Mycobacterium tuberculosis (Mtb)-driven lipid accumulation is intricately associated with the progression of tuberculosis (TB) disease. Although several studies elucidating the mechanisms for lipid droplet (LD) biosynthesis exist, we provide evidence for the significance of their regulated turnover via macroautophagy/autophagy during Mtb infection. We demonstrate that Mtb utilizes EGFR (epidermal growth factor receptor) signaling to induce the expression of the histone acetylation reader, BRD4 (bromodomain containing 4). The EGFR-BRD4 axis suppresses lipid-specific autophagy, and hence favors cellular lipid accumulation. Specifically, we found that pharmacological inhibition or knockdown of Egfr or Brd4 enhances autophagic flux and concomitantly decreases cellular LDs that is otherwise maintained at a significant level in chloroquine-treated or Atg5 knocked down autophagy-compromised host cells. In line with the enhanced lipophagy, we found that loss of EGFR or BRD4 function restricts mycobacterial burden that is rescued by external replenishment with oleic acid. We also report that the EGFR-BRD4 axis exerts additional effects by modulating pro-angiogenic gene expression and consequently aberrant angiogenesis during mycobacterial infection. This is important in the context of systemic Mtb dissemination as well as for the efficient delivery of anti-mycobacterial therapeutics to the Mtb-rich core of TB granuloma. Finally, utilizing an in vivo mouse model of TB, we show that pharmacological inhibition of EGFR and BRD4 compromises LD buildup via enhanced lipophagy and normalizes angiogenesis, thereby restricting Mtb burden and rescuing mice from severe TB-like pathology. These findings shed light on the novel roles of BRD4 during Mtb infection, and its possible implication in potentiating anti-TB responses.
Abbreviations: ATG5: autophagy related 5; BRDs: bromodomain containing; COL18A1: collagen type XVIII alpha 1 chain; EGFR: epidermal growth factor receptor; EP300: E1A binding protein p300; KDR: kinase insert domain receptor; KLF5: Kruppel like factor 5; LDs: lipid droplets; MAP1LC3B: microtubule associated protein 1 light chain 3 beta; Mtb: Mycobacterium tuberculosis; PECAM1: platelet and endothelial cell adhesion molecule 1; SQSTM1/p62: sequestosome 1; TB: tuberculosis; THBS1: thrombospondin 1; VEGF: vascular endothelial growth factor
Introduction
Mycobacterium tuberculosis (Mtb) reprograms host cellular processes to the benefit of its pathogenesis. Among the various strategies, accumulation of lipids in the form of lipid droplets (LDs) forms an essential determinant of Mtb survival. The neutral lipids and cholesterol esters harbored in the LDs provide a source of nutrition, regulate immune responses, and define critical aspects such as Mtb dormancy and reactivation [Citation1–3]. Therefore, it is imperative to understand the mechanisms underlying the formation of such lipid-laden foam cells, ranging from the transcriptional control of LD genes to processes that regulate lipid turnover such as autophagy.
Macroautophagy/autophagy is a highly regulated cell homeostatic process that has been shown to be co-opted by several pathogens, including Mtb, for their survival. A wide array of factors have been associated with the inhibition of this pathogen-clearance mechanism, including signaling pathways and miRNAs [Citation4–7]. Emerging studies offer strong evidence for the implication of distinct epigenetic factors in the realm of tuberculosis (TB) infection. Histone methyl transferases (such as EZH2, KMT5A [Citation8,Citation9]), histone demethylases (KDM6B [Citation10]), histone acetyl transferases (such as KAT5 [Citation11]) and histone deacetylases (SIRT1 [Citation12]) have been associated with the modulation of discrete cellular phenotypes that dictate Mtb survival. However, the identification of the readers of such epigenetic marks that may contribute to pivotal events in the pathogenesis of Mtb has been obscure. In this perspective, BRD (bromodomain containing) proteins, recognizing specific histone acetylation marks to govern gene expression, have gained relevance in several physiological and pathological processes. Among the different BRDs (BRD2, BRD3, BRD4 and BRDT), rapidly accumulating evidence have shown the implication of BRD4 in modulating innate immune responses during infections with L. monocytogenes, herpes simplex virus, human papillomavirus as well as in murine models of induced inflammation and endotoxin shock [Citation13–15]. In principle, the two bromodomains of BRD4 bind to two acetylated moieties on histone and/or non-histone proteins, including transcription factors, and recruit the necessary transcription machinery for modulating gene expression [Citation16]. To our interest, BRD4 has been reported to directly regulate lipogenesis [Citation17] and is also known to suppress autophagy [Citation18]. With this premise, we focused on elucidating the contribution of BRD4 in effectuating lipid accumulation during Mtb infection and its subsequent impact on TB pathogenesis.
At the systemic level, the granuloma forms as a result of structured congregation of immune cells at the site of infection that depicts a characteristic TB lesion [Citation19]. Interestingly, the core of the granuloma is generally abundant in LDs and displays a hypoxic environment [Citation20]. This hypoxia is known to drive angiogenic mechanisms in TB granulomas [Citation21]. Numerous studies pertaining to transcriptomics and serum analyses from Mtb-infected sources have identified the capacity of mycobacterial infection to induce angiogenic factors such as VEGFA (vascular endothelial growth factor A) and mediate blood vessel formation [Citation22]. However, a recent report demonstrated that TB-associated angiogenesis leads to the formation of inefficient blood vessels that strategically limit the access of immune cells and anti-TB therapeutics to the Mtb-harboring granuloma core [Citation23]. In this regard, the potential of anti-VEGF therapy and matrix remodelers have been harnessed for normalizing vasculature to enhance the delivery of small molecules into TB lesions [Citation24,Citation25]. These evidences emphasize on the importance of unraveling the intricate mechanisms and factors that may govern the progression of TB-associated angiogenesis.
We found that the epigenetic reader BRD4 is induced by Mtb in an EGFR (epidermal growth factor receptor) signaling-dependent manner, and it differentially regulates the expression of angiogenic genes, leading to ineffective angiogenesis. We also demonstrate the implication of EGFR and BRD4 in Mtb-driven LD generation and their sustenance in host cells via inhibition of lipophagy. Together, we report for the first time that Mtb utilizes EGFR-induced BRD4 to co-opt host cellular and immunological processes for successful pathogenesis. In corroboration, loss of function of EGFR or BRD4 was found to restore vasculature in the lung tissue while augmenting lipophagy and hence inhibiting LD accumulation at the cellular level. These outcomes directly assist in restricting Mtb burden in vitro and in vivo; thereby protecting mice from developing severe TB pathology in an in vivo therapeutic mouse model of TB. Besides, the current study highlights the pertinence of specific inhibitors of EGFR and BRD4 as probable adjuvants for anti-TB therapy.
Results
Mycobacteria-induced BRD4 facilitates TB pathogenesis in vivo
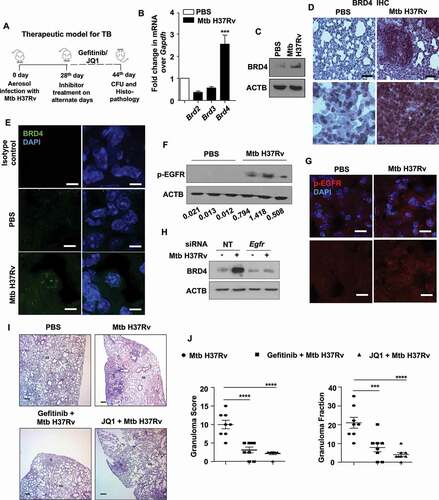
A therapeutic model of TB infection was utilized to study the effect of EGFR-BRD4 axis during Mtb pathogenesis in mice (). The transcript level of the BRD family members was assessed in the lungs of mice aerosol infected with Mtb H37Rv for 44 d, and among the tested, a significantly enhanced expression of BRD4 was observed (). In line, examination of the protein level of BRD4 by immunoblotting and immunofluorescence imaging in mouse peritoneal macrophages infected in vitro with Mtb H37Rv displayed its higher expression in infected samples compared to the uninfected controls (Fig. S1A and S1B). Corollary to this, the lung homogenates obtained from mice infected in vivo with Mtb H37Rv expressed BRD4 at an elevated level (), as was also confirmed in infected lung tissues by immunohistochemistry and immunofluorescence (,E). We next aimed to delineate the mechanism by which Mtb drives the expression of BRD4. Evidence from literature support the intricate correlation of BRD4 with EGFR in cellular processes relating to glioblastoma and ovarian cancers [Citation26,Citation27]. A study also identifies EGFR as a potential therapeutic target for TB [Citation27]. Therefore, we hypothesized if EGFR utilizes BRD4 in the realm of Mtb pathogenesis. To this end, first, we demonstrated that EGFR pathway is activated by Mtb H37Rv in vitro and in vivo (,G and S1C-E). siRNA-mediated knockdown of Egfr as well as its pharmacological inhibition using gefitinib compromised Mtb H37Rv-induced BRD4 expression, suggesting the possible implication of EGFR in driving BRD4-modulated immune functions during Mtb infection (, S1F and S1G). These findings prompted us to assess the relevance of Mtb-dependent epigenetic reader BRD4 and the receptor tyrosine kinase EGFR in an in vivo mouse model of tuberculosis (TB) (as depicted in ). Utilizing respective pharmacological inhibitors gefitinib and JQ1, we found that perturbation of EGFR or BRD4 significantly compromises the formation of hallmark TB granulomatous lesions in the lungs of treated mice, as indicated by granuloma fraction and total granuloma score (,J and S1H: arrows indicate granulomatous lesions). With these lines of evidence, we first report the EGFR-dependent expression of BRD4 during Mtb infection and highlight their crucial contribution in the pathogenesis of TB disease.
Figure 1. Epigenetic reader, BRD4, supports TB pathogenesis in vivo. (A) Schematic for the in vivo mouse TB model and therapeutic treatment regimen. (B) Lung homogenates from mice aerosol infected with Mtb H37Rv for 44 d were assessed for the expression of BRD family genes by qRT-PCR. (C-E) Lungs of mice infected with Mtb H37Rv for 44 d were assessed for the protein level expression of BRD4 by (C) immunoblotting in lung homogenates; (D) immunohistochemistry in infected lung sections and (E) by immunofluorescence imaging on infected lung cryosections. (F and G) The activation of EGFR was assessed (F) in the lung homogenates of 44 d-Mtb H37Rv-infected mice by immunoblotting, and (G) in the infected lung cryosections by immunofluorescence microscopy. (H) Mouse peritoneal macrophages were transfected with NT or Egfr siRNA and infected with Mtb H37Rv for 12 h. Whole cell lysates were assessed for BRD4 expression by immunoblotting. (I and J) H and E staining of lung sections of uninfected/infected/and inhibitor-treated mice (representative image, I; 40X; scale bar: 200 μm; G = granulomatous lesion; AS = alveolar space) and the respective quantification of granuloma score (J, left panel) and granuloma fraction (J, right panel). All qRT-PCR data represents mean±S.E.M. and immunoblotting data is representative of three independent experiments. At least 3 mice were utilized for each analysis derived from lung tissues obtained following therapeutic TB model. Granuloma score and fraction were evaluated in total 8 mice from two independent experiments. Med, medium; *** p < 0.001; ****, p < 0.0001 (Student’s t-test in B; One-way ANOVA in J; GraphPad Prism 8.0); scale bar for IF: 10 μm; Immunohistochemistry (IHC) magnification, 20X (upper panel; scale bar: 300 μm) and 40X (lower panel). ACTB was utilized as loading control.
EGFR-BRD4 axis regulates autophagy during mycobacterial infection
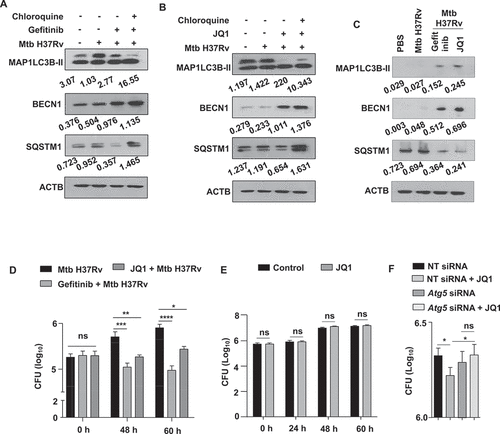
To elucidate the mechanism that enables EGFR-driven BRD4 to support Mtb infection, we initiated by assessing their contribution to autophagy as both have been independently reported as inhibitors of the process [Citation28,Citation29], and Mtb is also distinctly known to modulate its host to achieve the same [Citation30,Citation31]. Firstly, we analyzed a panel of autophagy-related genes for their transcriptional dependency on EGFR and BRD4 during Mtb H37Rv infection, without any significant correlation being found (Fig. S2A and S2B). However, we observed that pre-treatment of peritoneal macrophages with gefitinib or JQ1 augmented the autophagic flux, as may be appreciated with the accumulation of cleaved MAP1LC3B-II (microtubule associated protein 1 light chain 3 beta) and SQSTM1/p62 (sequestosome 1) in the presence of chloroquine (inhibitor of lysosomal acidification and cargo clearance) (,B). Corollary results were obtained in vivo from the lung homogenates of infected mice treated with respective EGFR and BRD4 inhibitors. Loss of SQSTM1 and accumulation of BECN1 and MAP1LC3B-II from gefitinib- and JQ1-treated lungs indicated an active autophagy (). Consequent to the augmented autophagy, we observed the mycobacterial burden to reduce in the presence of EGFR and BRD4 inhibitors (). Further, JQ1-dependent mycobacterial clearance could be reverted by blocking autophagy via Atg5 (autophagy related 5) knockdown (). We also confirmed that JQ1-mediated restriction of Mtb H37Rv burden in macrophages does not arise from any direct anti-mycobacterial activity of JQ1 by monitoring the growth kinetics and CFU of Mtb H37Rv cultured in the presence of JQ1 ( and S2C). These results indicate the possible role for host EGFR-BRD4 axis in favoring mycobacterial survival by suppressing the host cell function of autophagy.
Figure 2. EGFR-BRD4 axis suppresses autophagic flux. (A and B) Mouse peritoneal macrophages were infected Mtb H37Rv. Following 48 h, infected macrophages were treated with (A) gefitinib or (B) JQ1 for 12 h. Cells were then treated with chloroquine for 4 h where indicated, and whole cell lysates were analyzed for autophagy flux by immunoblotting. (C) The status of the indicated autophagy markers was assessed in the lung homogenates from the concerned sets of infected and treated mice (n = 3 mice in each group). (D) Murine peritoneal macrophages were infected with Mtb H37Rv for 4 h, extracellular Mtb H37Rv were washed and cells were incubated with the indicated inhibitors for 48 h and 60 h. Cells were lysed and plated on 7H11 to estimate intracellular Mtb H37Rv burden. (E) Mid log phase Mtb H37Rv cultures were sub-cultured in the presence or absence of JQ1 and plated on 7H11 medium to enumerate corresponding CFU. (F) Mouse peritoneal macrophages were transfected with NT or Atg5 siRNA as indicated. Transfected cells were infected with Mtb H37Rv for 60 h in the presence or absence of JQ1 and assessed for intracellular mycobacterial burden. NT, non-targeting; CFU, colony forming units; *, p < 0.05; **, p < 0.01; *** p < 0.001; ****, p < 0.0001; ns, not significant (Student’s t-test in D, One-way ANOVA in E; GraphPad Prism 5.0, 9.0). ACTB was utilized as loading control.
EGFR-dependent BRD4 couples Mtb-induced lipid accumulation and autophagy
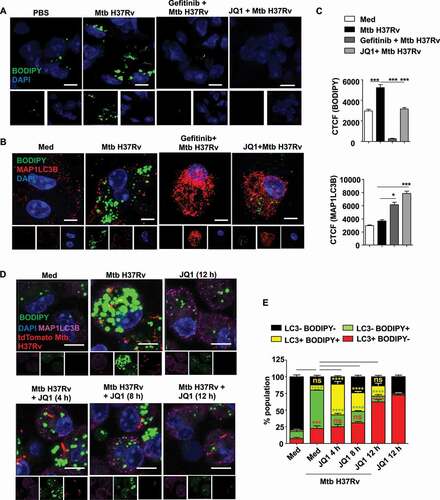
Literature evidence shows that BRD4 inhibition-induced autophagy does not mediate pathogen clearance, thereby suggesting the xenophagy arm to remain unaffected [Citation18]. However, our observation clearly demonstrates a compromised Mtb burden in the presence of JQ1, that is lost upon Atg5 knockdown (), indicating toward the possible loss of survival factors for Mtb via autophagy. One of the key components of Mtb pathogenesis is the formation of LDs that provide a source of nutrition and a secure niche for the bacterium [Citation1,Citation2]. Since lipids are among the various specified cargoes for autophagy [Citation32], we assessed the status of LDs in the presence of EGFR and BRD4 inhibitors. We found that Mtb-driven LD accumulation (BODIPY 493/503) is compromised in the lung sections of mice treated with inhibitors of EGFR and BRD4 (). Further, the premise that LD accumulation and autophagy are highly interdependent processes led us to speculate the prowess of Mtb to utilize EGFR-dependent BRD4 in regulating autophagy, and in turn LD accumulation. In vitro analysis of the possible interaction by co-staining for LDs (using BODIPY 493/503) and autophagy (using MAP1LC3B immunostaining) yielded an inverse correlation between the two processes (,C). Further investigation revealed that the LDs associate with MAP1LC3B (autophagy marker) as early as 4 h of JQ1 treatment, suggesting the utilization of autophagy as a readily available machinery for the turnover of lipids (,E). Importantly, a screen for various lipid biosynthesis (Dgat1, Dgat2, Fasn), uptake (Cd36), LD coating (Plin2) and degradation (Lpl, Lipa) markers highlighted critical LD accumulation genes (Cd36 and Plin2) to be expressed in EGFR-BRD4-dependent manner (Fig. S2D), indicating their sustained inhibition via transcriptional downregulation. This set of results demonstrate the EGFR-BRD4-dependent interaction of the processes of lipid accretion and autophagy.
Figure 3. EGFR-dependent BRD4 couples Mtb-induced lipid accumulation and autophagy. (A) Lipid accumulation was observed in the lung cryosections of mice infected with Mtb H37Rv, and treated with gefitinib or JQ1 as indicated, using BODIPY 493/503 staining (n = 3 mice in each group). (B and C) Murine peritoneal macrophages were pre-treated with gefitinib and JQ1 followed by 48 h infection with Mtb H37Rv. Lipid droplets were stained with BODIPY 493/503 and MAP1LC3B was immunostained and observed by confocal microscopy; (B) representative images and (C) the respective quantification. (D and E) Murine peritoneal macrophages were infected with tdTomato Mtb H37Rv for a duration of 60 h, with JQ1 treatment for 4 h, 6 h or 12 h prior to harvest, as indicated. Lipid accumulation and autophagy were assessed using BODIPY 493/503 and MAP1LC3B immunofluorescence, respectively; (D) representative image and (E) quantification. Med, medium; CTCF, corrected total cell fluorescence; *, p < 0.05; *** p < 0.001, ****, p < 0.0001; ns, not significant (one-way ANOVA in C; GraphPad Prism 5.0 & two-way ANOVA in E; GraphPad Prism 8.0); scale bar: 5 μm.
Lipophagy is subdued along EGFR-BRD4 axis during Mtb infection
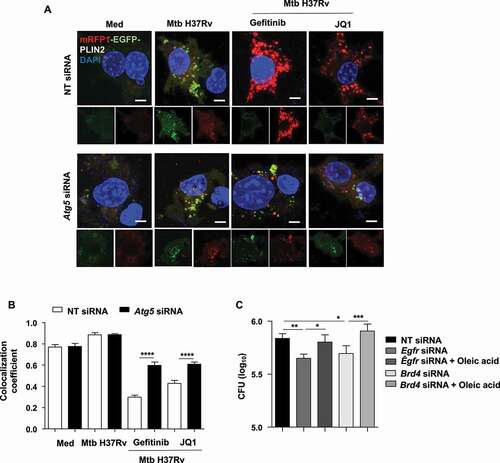
With our observations on inversely correlated LDs and autophagy and the colocalization of LDs with the autophagy marker MAP1LC3B, we investigated the possible clearance of lipids via the autophagic system upon gefitinib and JQ1 treatment by blocking autophagy with chloroquine (Fig. S3A and S3B) or by knocking down the key autophagy player Atg5 ( and S3C) [Citation33]. We found that compromising autophagic machinery reverses gefitinib- and JQ1-mediated reduction in LDs during Mtb infection, indicating toward lipophagy. The same was also observed in cells wherein Egfr and Brd4 were knocked down alongside Atg5 ( and S3D). This was further validated using PLIN2-traffic light construct (mRFP1-EGFP-PLIN2) that forms a direct determinant of the lipophagy flux. We observed an accumulation of mRFP1-EGFP-PLIN2 in Mtb-infected cells, depicting LD formation. However, treatment with gefitinib and JQ1 showed only an increased RFP signal, thereby confirming an enhanced flux of the traffic light construct through the autophagy system leading to quenched acidic pH sensitive-GFP fluorescence (,B). Since lipids form an essential entity for the survival of Mtb [Citation1,Citation2], we propose that the observed restriction in Mtb burden in the presence of gefitinib and JQ1 () may correspond to the lipophagy-active state of the host cells. To this end, we assessed mycobacterial burden in Egfr- and Brd4-knocked down macrophages, supplemented with an external source of lipids (oleic acid). We found that providing external lipids partially rescued Mtb burden (), suggesting lipophagy to be a possible mechanism by which inhibition of EGFR-BRD4 axis limits mycobacterial burden. Together, these results demonstrate the ability of Mtb to repurpose EGFR and BRD4 to its survival benefit by inducing LD genes as well as by subduing lipophagy. Next, we aimed to determine the effect of these signaling components on Mtb pathogenesis in vivo. Lipid accumulation deregulates various homeostatic processes and is often associated with hypoxic microenvironments and the production of pro-angiogenic factors that eventually lead to aberrant blood vessel formation [Citation34], a feature critical for defining the progression of TB disease and for the dissemination of Mtb [Citation35]. In line, during Mtb infection, LD-rich foam cells majorly localize in the hypoxic core of the granuloma [Citation36,Citation37], thus offering a crucial link between LDs and blood vessel formation, and thereby prompting us to evaluate the contribution of EGFR-BRD4 pathway in TB-associated angiogenesis.
Figure 4. Inhibition of EGFR-BRD4 induces lipophagy. (A) Murine peritoneal macrophages were transfected with NT or Atg5 siRNA. The transfected cells were infected for 48 h with tdTomato Mtb H37Rv followed by 12 h treatment with gefitinib or JQ1 and assessed for lipophagy by staining for LDs with BODIPY 493/503. (B) Mouse peritoneal macrophages were transfected with NT, Egfr or Brd4 siRNAs in the presence or absence of Atg5 siRNA. Transfected cells were infected with Mtb H37Rv for 60 h and assessed for lipid droplet accumulation (BODIPY 493/503) and autophagy (MAP1LC3B) by immunofluorescence. Med, Medium; NT, non-targeting; Scale bar: 5 μm.
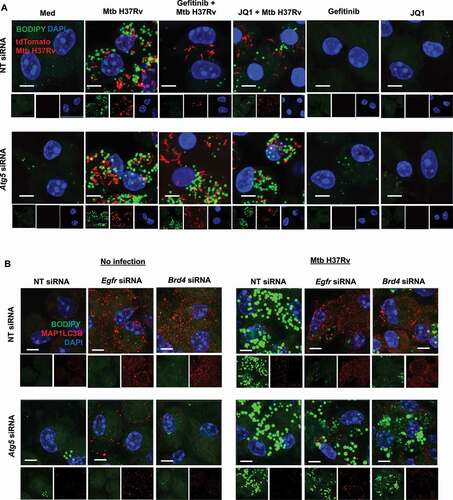
Figure 5. Inhibition of EGFR-BRD4 leads to mycobacterial clearance via LD depletion. (A and B) RAW 264.7 macrophages were transfected with mRFP1-EGFP-PLIN2 construct using Lipofectamine 3000 reagent, along with NT or Atg5 siRNA. The transfected cells were infected for 48 h with Mtb H37Rv followed by 12 h treatment with gefitinib or JQ1 and assessed for lipophagy by analyzing the colocalization of RFP and GFP signals; (A) representative images; (B) respective quantification. (C) Mouse peritoneal macrophages were transfected with NT or Egfr or Brd4 siRNA, in the presence or absence of oleic acid, and subsequently infected with Mtb H37Rv for 60 . Cells were lysed and plated on 7H11 medium to assess mycobacterial burden. Med, Medium; NT, non-targeting; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 (one-way ANOVA, GraphPad PRISM 9.0).
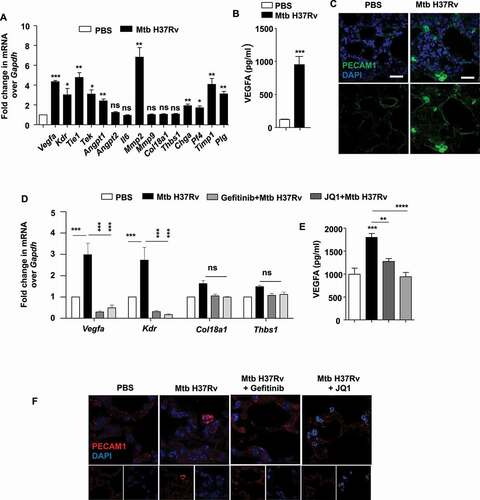
BRD4 effectuates Mtb-driven angiogenesis
The formation of blood vessels occurs from the concerted action of several pro- and anti-angiogenic genes. Firstly, we inspected a panel of such genes for their differential regulation during Mtb infection in vivo. We found a significantly enhanced expression of pro-angiogenic (Vegfa, Kdr, Tie1, Tek, Mmp2) and anti-angiogenic makers in the lungs of mice infected in vivo with Mtb H37Rv (). Substantiating the same, an elevated protein level of the pro-angiogenic factor VEGFA was observed in the lung homogenates of infected mice (), with concomitantly augmented PECAM1 (platelet and endothelial cell adhesion molecule 1) staining in lung tissues demarcating endothelial cells constituting the blood vessels (). With this premise, we utilized the pharmacological inhibitor of BRD4 (JQ1) and Brd4 siRNA and found that the expression of pro-angiogenic genes (such as Vegfa) was strongly dependent on BRD4 function (Fig. S4A-C); with no evident effect on the anti-angiogenic markers. In line with these findings, a significant upregulation of Vegfa and Kdr transcripts was observed in RAW 264.7 macrophages overexpressing BRD4 WT construct, without any effect on the levels of the anti-angiogenic markers Thbs1 (thrombospondin 1) and Col18a1 (collagen type XVIII alpha 1 chain) (Fig. S4D). Further, we found EGFR-BRD4 axis to regulate the expression of pro-angiogenic genes in the lung homogenates and tissue sections obtained from mice subjected to our in vivo therapeutic model of TB () by qRT-PCR, ELISA and immunofluorescence assays (–F). These results indicate an intricate function of BRD4 in differentially regulating angiogenic genes during Mtb infection. Importantly, this aligns with studies that correlate lipid accumulation with an increased expression of pro-angiogenic factors such as VEGF and HGF (hepatocyte growth factor) [Citation38].
Figure 6. BRD4 modulates angiogenic genes during mycobacterial infection. (A) The transcript levels of the indicated angiogenic genes were assessed in the lung homogenates of mice infected for 44 d with Mtb H37Rv. (B) The protein level of VEGFA was analyzed in Mtb H37Rv-infected lung homogenates by ELISA. (C) The expression of PECAM1 was ascertained by immunofluorescence imaging of infected lung sections. (D) Lung homogenates of infected mice treated with gefitinib or JQ1 was analyzed for the expression of the indicated angiogenic markers by qRT-PCR. (E) The protein level of VEGFA was assessed in the lung homogenates of the indicated groups of mice by ELISA. (F) The expression of PECAM1 was assessed in the lung cryosections of Mtb H37Rv-infected and gefitinib/JQ1-treated mice. At least 3 sets of mice were utilized for each analysis derived from lung tissues obtained following therapeutic TB model and qRT-PCR data represents mean±S.E.M. from five sets of mice. Med, medium; *, p < 0.05; **, p < 0.005; ***, p < 0.001; ****, p < 0.0001 (Students t-test in A, B and one-way ANOVA in D, E; GraphPad Prism 5.0 and 8.0); ns, not significant; scale bar: 10 μm.
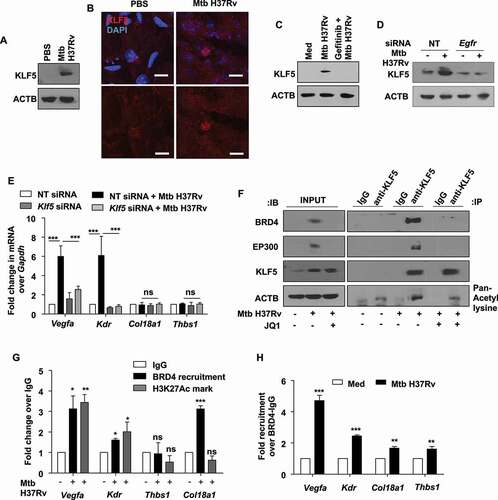
KLF5 mediates BRD4-dependent modulation of angiogenic genes
The transcriptional regulation of Mtb-induced Vegfa and Kdr by the epigenetic mark reader BRD4 requires BRD4-driven assembly of transcription machinery at the concerned promoters. As introduced, BRD4 influences gene expression by interacting with acetylated moieties of histones or transcription factors. Among several possible targets, certain pieces of evidence suggest that KLF5 (Kruppel like factor 5) is activated by EP300 (E1A binding protein p300)-mediated acetylation [Citation39]; and is involved in the modulation of Vegfa expression and angiogenesis [Citation40]. We found that KLF5 is expressed in the lungs of mice infected with Mtb H37Rv (,B), and in vitro analysis indicated its expression to depend on Mtb-activated EGFR signaling (,D). Loss of KLF5 by siRNA-mediated knockdown compromised the transcript-level expression of the concerned pro-angiogenic markers, Vegfa and kdr (). Akin to BRD4, KLF5 also showed no impact on the expression of anti-angiogenic genes Thbs1 and Col18a1. With these observations, we surmised the intersection of KLF5 and BRD4 to shape Mtb-induced angiogenic dysregulation. In this context, KLF5 was found to interact with EP300 and undergo acetylation during Mtb infection. Further, it was observed that Mtb favored the interaction of BRD4 and acetylated KLF5 (). Congruently, we could observe an enhanced recruitment of BRD4 and associated H3K27 acetylation mark at KLF5-binding regions on the promoters of the indicated pro-angiogenic markers, Vegfa and Kdr (). Sequential ChIP showing high co-occupancy of the promoters of Vegfa and Kdr with BRD4 and KLF5 further substantiated our findings. Such co-occupancy was partly compromised on the promoters of anti-angiogenic genes, Col18a1 and Thbs1 (). These results indicate the coordinated action of KLF5 and BRD4 in differentially regulating Mtb-induced angiogenic progression. Alongside, we assessed the possible contribution of KLF5 to lipophagy. We found that the loss of KLF5 function disrupts lipid droplet accumulation, however, majorly independent of autophagic flux. This is because knocking down Atg5 did not restore Klf5 knockdown-dependent reduction in LDs (Fig. S5A and S5B), indicating possible alternate mechanisms of lipid accumulation via KLF5 and the specificity of EGFR-BRD4 axis in subduing lipophagy in this context.
Figure 7. Mtb-dependent KLF5 assists BRD4-mediated regulation of angiogenic markers. (A and B) KLF5 protein expression was assessed (A) in the lung homogenates of 44 d-Mtb H37Rv-infected mice by immunoblotting and (B) in the lung cryosections of infected mice by immunofluorescence imaging (n = 3 mice in each group). (C) Murine peritoneal macrophages were pre-treated with EGFR inhibitor gefitinib for 1 h, followed by 12 h infection with Mtb H37Rv; and whole cell lysates were assessed for the expression of KLF5 by immunoblotting. (D) Mouse peritoneal macrophages were transfected with NT or Egfr siRNA and infected with Mtb H37Rv for 12 h. Whole cell lysates were assessed for KLF5 expression by immunoblotting. (E) Murine peritoneal macrophages were transfected with Klf5 siRNA or NT siRNA for 24 h. Transfected cells were infected with Mtb H37Rv for 12 h and assessed for the expression of the indicated angiogenic genes. (F) Murine peritoneal macrophages were infected with Mtb H37Rv for 12 h and assessed for the interaction of BRD4, EP300 and KLF5 and KLF5 acetylation by immunoprecipitation assay. (G) The recruitment of BRD4 and corresponding H3K27Ac marks were analyzed on the promoters of the indicated genes (at KLF5 binding regions) by ChIP. (H) Sequential ChIP was performed to access the co-occupancy of the indicated promoters by KLF5 and BRD4 in uninfected and Mtb H37Rv-infected peritoneal macrophages. All qRT-PCR data represents mean±S.E.M. and immunoblotting data is representative of three independent experiments. Med, medium; *, p < 0.05; **, p < 0.005; *** p < 0.001 (Students t-test in H and one-way ANOVA in E, G; GraphPad Prism 5.0); ns, not significant; scale bar: 5 μm. ACTB was utilized as loading control.
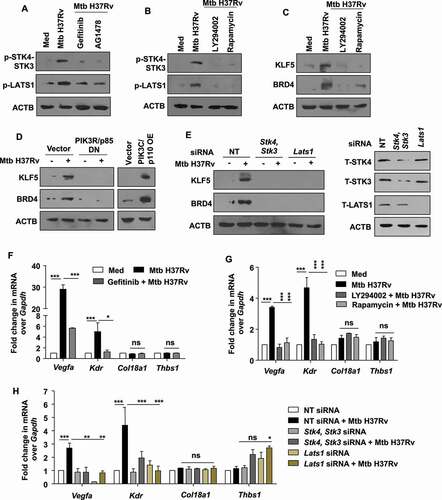
EGFR-PI3K-HIPPO axis drives BRD4-dependent angiogenic deregulation during Mtb infection
Having observed the interaction of BRD4 and KLF5 to modulate angiogenic genes, we delineated the mechanism adopted by Mtb-driven EGFR pathway to regulate the expression of the aforesaid factors. EGFR utilizes several adaptors and signal transducers to alter cellular gene expression [Citation41,Citation42]. In an attempt to identify novel intermediates that lead to the expression of BRD4, we speculated the implication for HIPPO pathway as HIPPO is reported to be repurposed by Mtb for the expression of chemokines and inflammatory cytokines [Citation43]; and has also been independently reported as a regulator of angiogenesis [Citation44,Citation45]. In this context, we found that Mtb-induced EGFR signaling regulated HIPPO pathway, as perturbation of EGFR through pharmacological inhibitors compromised the ability of Mtb to effectuate HIPPO signaling activation in Mtb H37Rv-infected macrophages (). Consisting of a series of serine threonine kinases, STK4 (serine/threonine kinase 4)-STK3 cannot be directly activated by the receptor tyrosine kinase, EGFR. Canonical downstream EGFR signaling adaptors PI3K and MTOR [Citation46] were found to regulate HIPPO; as Mtb failed to activate HIPPO pathway components STK4-STK3 and LATS1 (large tumor suppressor kinase 1) in the presence of respective pharmacological inhibitors LY294002 (a PI3K and PtdIns3K inhibitor) and rapamycin (MTOR inhibitor) (). Further, we found BRD4 and KLF5 to be expressed along the PI3K-MTOR-HIPPO signaling axis; as perturbation of either component led to a compromised expression of BRD4 and KLF5 in the presence of Mtb H37Rv infection (–E).
Figure 8. EGFR-PI3K-HIPPO axis regulates angiogenesis markers during mycobacterial infection. (A) Murine peritoneal macrophages were pre-treated with gefitinib and AG1478, followed by 1 h infection with Mtb H37Rv. Whole cell lysates were assessed for the activation of the indicated HIPPO pathway components by immunoblotting. (B) Peritoneal macrophages were pre-treated with LY294002 (PI3K and PtdIns3K inhibitor) or rapamycin (MTOR inhibitor), followed by 1 h infection with Mtb H37Rv and assessed for the activation of Hippo pathway by immunoblotting. (C) Peritoneal macrophages were pre-treated with LY294002 or rapamycin, followed by 12 h infection with Mtb H37Rv and assessed for the expression of KLF5 and BRD4 by immunoblotting. (D) RAW 264.7 macrophages were transfected with the indicated constructs (PIK3R/p85 DN, PIK3C/p110 OE) and transfected cells were infected with Mtb H37Rv for 12 h. Whole cell lysates were assessed for the expression of KLF5 and BRD4 by immunoblotting. (E) Peritoneal macrophages were transfected with Stk4 and Stk3 siRNA, Lats1 siRNAs or NT siRNA for 24 h followed by 12 h infection with Mtb H37Rv; and assessed for (E, left panel) the protein levels of KLF5, BRD4; and (E, right panel) knockdown of Stk4, Stk3 and Lats1, by immunoblotting. (F) The expression of the indicated angiogenic genes was analyzed in the murine peritoneal macrophages pre-treated with gefitinib for 1 h and followed by 12 h infection with Mtb H37Rv. (G) Peritoneal macrophages were pre-treated with LY294002 or rapamycin, followed by 12 h infection with Mtb H37Rv and assessed for the expression of the indicated angiogenesis genes by qRT-PCR. (H) Peritoneal macrophages were transfected with Stk4 and Stk3 siRNA, Lats1 siRNA or NT siRNA for 24 h followed by 12 h infection with Mtb H37Rv; and assessed for the expression of angiogenesis genes by qRT-PCR. All qRT-PCR data represents mean±S.E.M. and immunoblotting data is representative of three independent experiments. NT, non-targeting; Med, medium; DN, dominant-negative; OE, overexpression; *, p < 0.05; **, p < 0.005; *** p < 0.001; ns, not significant (one-way ANOVA in F-H; GraphPad Prism 5.0); ACTB was utilized as loading control.
Having the premise that EGFR integrates the activation of HIPPO signaling; its further role in Mtb-induced angiogenic deregulation was surmised. It was found that EGFR-PI3K-MTOR-HIPPO axis plays a critical role in defining angiogenic gene expression during Mtb H37Rv infection. Abrogation of EGFR-PI3K-MTOR using specific inhibitors; and the HIPPO pathway using siRNA-mediated knockdown; suppressed the ability of Mtb to drive Vegfa and Kdr expression (–H). Thus, the process of Mtb granuloma-associated angiogenesis was found to be finely regulated by BRD4 and KLF5 along the EGFR-PI3K-MTOR-HIPPO signaling axis.
Perturbation of EGFR and BRD4 restricts mycobacterial survival in vivo
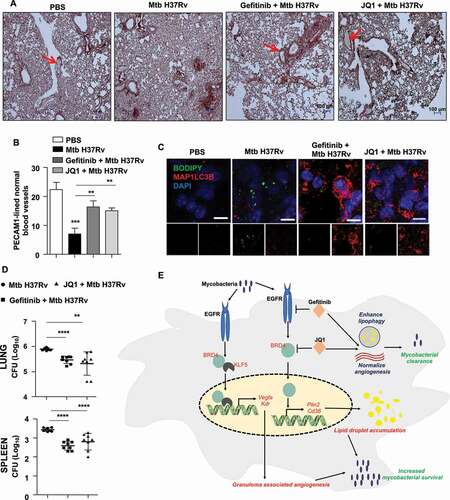
The observation that EGFR-dependent BRD4 governs the expression of angiogenic markers, LD accumulation and autophagy in vitro and in vivo; led us to identify their role in Mtb survival within the host by utilizing mouse model of TB infection (depicted in ). Firstly, we assessed the formation of PECAM1-lined normal blood vessels during Mtb infection. This is because excessive VEGFA production is associated with pathological angiogenesis characterized by constricted and blocked blood vessels. This feature is considered particularly beneficial for Mtb as inefficient blood flow through such aberrant vessels would keep immune cells and anti-mycobacterial therapeutics from reaching the Mtb-containing core of the granuloma. It was observed that PECAM1- stained endothelial cells that demarcate the lining of blood vessels displayed empty or blood-filled spaces in uninfected and inhibitor-treated mice, as indicated by the red arrows (). However, infection of mice with Mtb H37Rv led to concentrated PECAM1 staining, without the presence of any noticeable lumen, depicting abnormal and inefficient vasculature. Thus, treatment of mice with gefitinib or JQ1 restored blood vessel architecture as quantified in . Also, as speculated, inhibition of EGFR and BRD4 inhibited LD accumulation and augmented autophagy in the lung sections of infected mice (). With these findings, the overall relevance of the orchestrated induction of EGFR-BRD4 axis-defined angiogenesis, autophagy and LDs was assessed by analyzing the mycobacterial burden. Congruent to the resolution of hallmark TB granulomatous lesions observed in ,J, a reduction in mycobacterial burden was found in the lungs (, upper panel) and spleen (, lower panel) of infected mice that were treated with gefitinib or JQ1. This set of in vivo experiments indicates that EGFR-BRD4 axis plays a crucial role in regulating TB-associated angiogenesis and lipophagy, and subsequently Mtb survival within the host. Together, we propose EGFR and BRD4 inhibitors as potent anti-mycobacterial therapeutic adjuvants for their capacity to target multiple strategies adopted by Mtb for its persistence.
Figure 9. EGFR-BRD4 aids in mycobacterial survival. (A and B) Immunohistochemical analysis of PECAM1 expression in the lung sections of the indicated groups of mice and the respective quantification. PECAM1-lined blood vessels with blood-filled lumen are close representatives of normal vasculature (n = 3 mice in each group). (C) Lung cryosections from the indicated groups of mice were analyzed for autophagy and LD accumulation by MAP1LC3B immunostaining and BODIPY 493/503 staining, respectively (n = 3 mice in each group). (D) The indicated groups of mice were assessed for Mtb H37Rv CFU in the lungs (D, top panel) and spleen (D, bottom panel) (n = 8 mice in each group). (E) Model: Mycobacteria coopts EGFR-BRD4 axis to promote aberrant angiogenesis and suppress lipophagy to thrive within the host milieu. **, p < 0.01; *** p < 0.001; **** p < 0.0001 (one-way ANOVA in B, D; GraphPad Prism 9.0).
Discussion
In the current study, we showed that EGFR signaling-dependent BRD4 transcriptionally regulates the genes responsible for lipid accumulation and aids in LD buildup by actively suppressing autophagy. Although, the significance of autophagy and LDs have been individually detailed in various studies, their correlation during Mtb infection has remained understudied. One of the existing reports shows an inverse correlation of LDs and autophagy through the action of Mir33 and Mir33* on the components of the two pathways. However, it does not clearly demonstrate the intersection of these two processes [Citation47]. We provide the first evidence for the suppression of LD turnover via autophagy in Mtb-infected cells, and that this restriction requires the participation of EGFR and BRD4. Further, we believe that BRD4 might act in concert with the H3K27 demethylase KDM6B to effectuate the transcriptional control of LD genes by recognizing active acetylation marks conferred on H3K27 sites upon the already known removal of H3K27me3 by KDM6B from specific LD gene promoters [Citation9]. Importantly, we support the use of BRD4 inhibitors as anti-mycobacterial agents by virtue of their potential to limit pro-survival factors for Mtb, despite their reported non-association with pathogen-specific xenophagy. We demonstrated that the loss-of-function of EGFR and BRD4 induces lipophagy that depletes the host cells of the LDs essential for the persistence of Mtb. Consequently, supplementation with external lipid sources, such as oleic acid, partially restored mycobacterial burden. With these observations, we also find it imperative to emphasize that multiple parallel cellular pathways exist for lipid breakdown, including autophagy (lysosomal) and cytosolic enzyme-mediated degradation; each of which would contribute to lipid availability for Mtb survival. Thus, the overall impact and the segregation or inter-regulation of such cellular processes on mycobacterial pathogenesis would require further elucidation.
The central role of lipids does not limit to survival, dormancy and reactivation of Mtb. With the availability of literature evidence demonstrating the correlation of the deposition of foam cells and formation of cholesterol crystals with neo-angiogenesis in clinical manifestations such as atherosclerosis [Citation48], we sought to assess the molecular regulation of TB-associated blood vessel formation along the EGFR-BRD4 axis. We showed that the expression of pro-angiogenic markers, majorly Vegfa and Kdr, is dependent on EGFR and BRD4. Inhibition of this pathway compromises the expression of the concerned angiogenic genes and normalizes blood vessel formation. As as been reported that normalized vasculature leads to a better prognosis for TB, we proposed that specific inhibitors for EGFR and BRD4 may serve as potential adjuvants in amplifying the effectiveness of anti-TB therapies. This study provides mechanistic insights into the hitherto unknown regulatory circuits operating in host cells for effectuating TB-associated angiogenesis, thus widening our arsenal that may be targeted for therapeutic purposes..
Together, we first report that the epigenetic mark reader, BRD4, is exploited by Mtb to execute multifaceted immune evasion strategies. We presented the molecular mechanism of transcriptional regulation of angiogenesis; and the inter-dependency of LDs and autophagy during TB infection (). We found a distinctive impact of EGFR-BRD4 inhibition on Mtb burden and TB pathogenesis. Moreover, we did not rule out the implication of this reader in the modulation of any other Mtb-driven immune subversion strategies. For instance, BRD4 is well known for its ability to mediate nuclear factor kappa B (NFKB)-driven inflammatory responses in the context of several viral infections and septic shock. With the ability of mycobacteria to intercept a plethora of host immune pathways, including NFKB-mediated events, we believe that EGFR-BRD4 axis may contribute to additional immune cross-talks. Interestingly, genome-wide association studies have now started to identify the correlation of single nucleotide polymorphisms (SNPs) in the enhancer regions of gens with specific disease states. Such observations highlight the relevance of targeting epigenetic mechanisms, including BRD4 as one of the readers of such SNPs for the treatment of diseases [Citation49]. Alongside, host-directed therapeutics (HDTs) is garnering interest amid the dwindling development of novel antibiotics and rapid emergence of antibiotic resistance. In this light, we believe that strategies targeting these factors would provide rational and clinically relevant adjuncts for TB treatment.
Materials and methods
Cells and mice
Four- to six-weeks-old male and female BALB/c mice were utilized for all experiments. Mice were procured from The Jackson Laboratory (stock number 000651) and maintained at the Central Animal Facility (CAF) in Indian Institute of Science (IISc) under 12-h light and dark cycle. For in vitro experiments, mouse peritoneal macrophages were utilized. Briefly, mice were injected intraperitoneally with 8% Brewer’s thioglycolate (HiMedia, M019) and peritoneal exudates were harvested in ice cold PBS (HiMedia, M1866TL1006) after 4 d, seeded in tissue culture dishes. Adherent cells were utilized as peritoneal macrophages. RAW 264.7 mouse monocyte-like cell line was procured from the National Centre for Cell Science (NCCS; ATCC TIB-71). All cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Gibco, Thermo Fisher Scientific, 12100061) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco, Thermo Fisher Scientific, 10270106) and maintained at 37°C in 5% CO2 incubator.
Ethics statement
Experiments involving mice were carried out after the approval from Institutional Ethics Committee for animal experimentation. The animal care and use protocol adhered to were approved by national guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Government of India. Experiments with virulent mycobacteria (Mtb H37Rv) were approved by the Institutional Biosafety Committee.
Bacteria
Virulent strain of Mtb (Mtb H37Rv) was a kind research gift from Prof. Kanury Venkata Subba Rao (THSTI, India). Mycobacteria were cultured in Middlebrook 7 H9 medium (Difco, DF0713-17-9) supplemented with 10% OADC (oleic acid [Sigma-Aldrich, O1008], ALB [albumin; HiMedia, MB083], dextrose [HiMedia GRM077], CAT [catalase; HiMedia, RM446]). Single-cell suspensions of mycobacteria were obtained by passing mid log phase culture through 23-, 28- and 30-gauge needle 10 times each and used for infecting primary cells or RAW 264.7 macrophages at multiplicity of infection 10 (macrophage:mycobacteria, 1:10). The studies involving virulent mycobacterial strains were carried out at the biosafety level 3 (BSL-3) facility at Center for Infectious Disease Research (CIDR), IISc.
Reagents and antibodies
All general chemicals and reagents were procured from Sigma-Aldrich/Merck Millipore, HiMedia and Promega. Tissue culture plasticware was purchased from Jet Biofil, Tarsons India Pvt. Ltd. and Corning Inc. siRNAs were obtained from Dharmacon as siGENOME SMART-pool reagents against Klf5, Stk4, Stk3, Lats1, Atg5. Egfr and Brd4 siRNAs were purchased from Eurogentec. Oleic acid, HRP-tagged anti-ACTB (A3854), 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI, 10236276001), 3,3′-diaminobenzidine tetrahydrochloride hydrate (DAB, D5637), and anti-MAP1LC3B (L7543) were procured from Sigma-Aldrich. Anti-phospho-STK4-STK3 (3681), anti-STK4 (14946), anti-STK3 (3952), anti-phospho-LATS1 (8654), anti-LATS1 (3477), anti-BECN1 (3738), anti-SQSTM1 (5114), anti-phospho-EGFR (2231), anti-EGFR (2232), anti-EP300 (59240), anti-acetylated lysine (9441) and anti-acetyl H3K27 (4353) antibodies were purchased from Cell Signaling Technology. Anti-KLF5 antibody (PA5-27876) was purchased from Thermo Fisher Scientific, anti-PECAM1 (ab288364) and anti-BRD4 (ab75898) antibodies were procured from Abcam. VEGFA ELISA kit (900-K99) was obtained from PeproTech, USA. Lipofectamine 3000 (L3000015) was purchased from Thermo Fisher Scientific.
Treatment with pharmacological reagents
All the reagents were procured from Calbiochem, Sigma-Aldrich, PeproTech and Cayman Chemicals. Primary macrophages treated with the following reagents 1 h prior to infection with Mtb H37Rv unless otherwise stated: Gefitinib (Cayman Chemicals, 13166; 20 μM); JQ1 (Cayman Chemicals, 11187; 400 nM); LY294002 (Calbiochem, 440202; 50 μM), rapamycin (Calbiochem, 553210; 100 nM). Chloroquine (Sigma-Aldrich, C6628; 10 μM) was added 6 h prior to harvest.
Transient transfection studies
RAW 264.7 macrophages were transfected with the indicated constructs (PI3K overexpression [PIK3C/p110 OE], PI3K dominant-negative [PIK3R/p85 DN], BRD4 WT); or primary macrophages were transfected with 100 nM each of siGLO Lmna (lamin A/C; Horizon, D-001620-01), non-targeting siRNA or specific siRNAs with the help of polyethyleneimine (PEI; Sigma-Aldrich, 764,604) or Lipofectamine 3000 for 8 h; followed by 24 h recovery. 70–80% transfection efficiency was observed by counting the number of siGLO Lmna-positive cells in a microscopy field using fluorescence microscopy. Transfected cells were subjected to the required infections/treatments for the indicated time points and processed for analyses.
RNA isolation and quantitative real time PCR (qRT-PCR)
Treated samples were harvested in TRIzol (Sigma-Aldrich, T9424) and incubated with chloroform for phase separation. Total RNA was precipitated from the aqueous layer. Equal amount of RNA was converted into cDNA using First Strand cDNA synthesis kit (Applied Biological Materials Inc., G236). The cDNA thus obtained was used for SYBR Green (Thermo Fisher Scientific, F416) based quantitative real time PCR analysis for the concerned genes. Gapdh was used as internal control gene. Primer pairs used for expression analyses are detailed in Table S1.
Immunoblotting
Cells post treatment and/or infection were washed with PBS. Whole cell lysate was prepared by lysing in RIPA buffer (50 mM Tris-HCl, pH 7.4, 1% NP40 [Sigma-Aldrich, 74,385], 0.25% sodium deoxycholate [Sigma-Aldrich, D6750], 150 mM NaCl, 1 mM EDTA, 1 mM PMSF [Sigma-Aldrich, 78,830], 1 μg/ml each of aprotinin [Sigma-Aldrich, A4529], leupeptin [Sigma-Aldrich, L2884], pepstatin [Sigma-Aldrich, P4265], 1 mM Na3VO4 [Sigma-Aldrich, S6508], 1 mM NaF [Sigma-Aldrich, S7920]) on ice for 30 min. Total protein from whole cell lysates was estimated by Bradford reagent (Sigma-Aldrich, B6916). Equal amount of protein was resolved using 12% SDS-PAGE and transferred onto PVDF membranes (Millipore, IPVH00010) by the semi-dry immunoblotting method (Bio-Rad). Nonfat dry milk powder (HiMedia, GRM1254) (5%) in TBST (20 mM Tris-HCl [Sigma-Aldrich, 10812846001], pH 7.4, 137 mM NaCl [Sigma-Aldrich, 746398, 0.1% Tween 20 [Sigma-Aldrich, P9416]) was used for blocking nonspecific binding for 60 min. After washing with TBST, the blots were incubated overnight at 4°C with primary antibody diluted in TBST with 5% BSA (HiMedia, MB083). After washing with TBST, blots were incubated with anti-rabbit IgG secondary antibody conjugated to HRP antibody (Jackson ImmunoResearch, 111–035-045) for 4 h at 4°C. The immunoblots were developed with enhanced chemiluminescence detection system (Perkin Elmer, NEL105001EA) as per the manufacturer’s instructions. For developing more than one protein at a particular molecular weight range, the blots were stripped off the first antibody at 60°C for 5 min using stripping buffer (62.5 mM Tris-HCl, pH 6.8, with 2% SDS [Sigma-Aldrich, L3771], 100 mM 2-mercaptoethanol [Sigma-Aldrich, M314A]), washed with TBST, blocked; followed by probing with the subsequent antibody following the described procedure. ACTB was used as loading control.
Immunoprecipitation assay
Immunoprecipitation assays were carried out following a modified version of the protocol provided by Millipore, USA. Treated samples were washed in ice-cold PBS and gently lysed in RIPA buffer. The cell lysates obtained were subjected to pre-clearing with BSA-blocked protein A beads (Bangalore Genei, LIA1M) for 30 min at 4°C and slow rotation. The amount of protein in the supernatant was quantified and equal amount of protein was used for pull down from each treatment condition; using Protein A beads pre-conjugated with the antibody of interest or isotype control IgG antibody. After incubation of the whole cell lysates with the antibody-complexed beads for 4 h at 4°C on slow rotation, the pellet containing the bead-bound immune complexes were washed with RIPA buffer twice. The complexes were eluted by boiling the beads in Laemmli buffer for 10 min. Bead-free samples were resolved by SDS-PAGE and the target interacting partners were identified by immunoblotting. Clean-Blot IP Detection Reagent (Thermo Fisher Scientific, 21230) was used for immunoblotting of the specific proteins.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were carried out using a protocol provided by Upstate Biotechnology and Sigma-Aldrich with certain modifications. Briefly, treated samples were washed with ice-cold PBS and fixed with 3.6% formaldehyde for 15 min at room temperature followed by inactivation of formaldehyde with 125 mM glycine. Nuclei were lysed in 0.1% SDS lysis buffer (50 mM Tris-HCl, pH 8.0, 200 mM NaCl, 10 mM HEPES, pH 6.5, 0.1% SDS, 10 mM EDTA, 0.5 mM EGTA, 1 mM PMSF, 1 μg/ml of each aprotinin, leupeptin, pepstatin, 1 mM Na3VO4 and 1 mM NaF). Chromatin was sheared using Bioruptor Plus (Diagenode, Belgium) at high power for 70 rounds of 30 s pulse ON and 45 s pulse OFF. Chromatin extracts containing DNA fragments with an average size of 500 bp were immunoprecipitated with KLF5 or BRD4 or H3K27Ac or rabbit preimmune sera complexed with protein A agarose beads (Bangalore Genei, LIA1M). Immunoprecipitated complexes were sequentially washed twice with Wash Buffer A, B and TE (Wash Buffer A: 50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 1 mM EDTA, 1% Triton X-100 [Sigma-Aldrich, T8787], 0.1% sodium deoxycholate, 0.1% SDS and protease/phosphatase inhibitors [1 μg/ml each of aprotinin [Sigma-Aldrich, A4529], leupeptin [Sigma-Aldrich, L2884], pepstatin [Sigma-Aldrich, P4265], 1 mM Na3VO4 [Sigma-Aldrich, S6508], 1 mM NaF [Sigma-Aldrich, S7920]; Wash Buffer B: 50 mM Tris-HCl, pH 8.0, 1 mM EDTA, 250 mM LiCl, 0.5% NP40, 0.5% sodium deoxycholate and protease/phosphatase inhibitors; TE: 10 mM Tris-HCl, pH 8.0, 1 mM EDTA) and eluted in elution buffer (1% SDS, 0.1 M NaHCO3). After treating the eluted samples with RNase A (Thermo Fisher Scientific, EN0531) and Proteinase K (Thermo Fisher Scientific, 25,530,049), DNA was purified and precipitated using phenol-chloroform-ethanol method. Purified DNA was analyzed by quantitative real time RT-PCR. All values in the test samples were normalized to amplification of the specific gene in Input and IgG pull down and represented as fold-change in modification or enrichment. All ChIP experiments were repeated at least three times. The list of primers is given in Table S2.
Sequential ChIP assay
The protocol for sequential ChIP was adopted from [Citation50,Citation51]. Briefly, the DNA fragments obtained from fixed cells following sonication (in lysis buffer; 1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.0) were immunoprecipitated with BRD4-complexed protein A beads. After first pull down, beads were washed with Re-ChIP Buffer (2 mM EDTA, 500 mM NaCl, 0.1% SDS, 1% NP40), followed by elution of DNA in Re-ChIP elution buffer (2% SDS, 15 mM DTT in TE) at 37°C for 30 min. The eluted DNA was subjected to subsequent round to immunoprecipitation with Protein-A beads pre-complexed with KLF5 or rabbit pre-immune sera. Immunoprecipitated complexes were sequentially washed with Wash Buffer A, B and TE (Wash Buffer A: 20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS and protease/phosphatase inhibitors; Wash Buffer B: 20 mM Tris-HCl, pH 8.0, 2 mM EDTA, 500 mM NaCl, 1% Triton X-100, 0.1% SDS and protease/phosphatase inhibitors; Wash Buffer C: 10 mM Tris-HCl, pH 8.0, 1 mM EDTA, 1% sodium deoxycholate, 1% NP40, 250 mM LiCl and protease/phosphatase inhibitors; TE: 10 mM Tris-HCl, pH 8.0, 1 mM EDTA and protease/phosphatase inhibitors) and eluted (0.1 M NaHCO3, 1% SDS), purified and subjected to qRT-PCR (as described previously). The fold-change of BRD4-KLF5 versus BRD4-IgG upon infection signified the co-occupancy of the two factors at concerned promoters. The list of primers is given in Table S2.
In vitro CFU analysis
Primary mouse macrophages were infected with Mtb H37Rv at MOI 1 for 4 h. Post 4 h, the cells were thoroughly washed with PBS to remove any surface adhered bacteria and medium containing amikacin (HiMedia, CMS644) (0.2 mg/ml) was added for 2 h to deplete any extracellular mycobacteria. After amikacin treatment, the cells thoroughly washed with PBS were taken for 0 h time point. A duplicate set was maintained in antibiotic-free medium for the indicated time points along with respective inhibitors, gefitinib and JQ1. Similar protocol was followed for macrophages knocked down for Atg5. Intracellular mycobacterial burden was enumerated by lysing macrophages with 0.06% SDS in 7H9 Middlebrook medium. Appropriate dilutions were plated on Middlebrook 7H11 agar plates supplemented with OADC. Total colony-forming units (CFUs) were counted after 21 d of plating.
Mtb growth kinetics in the presence of JQ1
Mtb H37Rv was grown to exponential phase (O.D. 0.6), then diluted to 0.1 O.D. and cultured in the presence of JQ1 (400 nM) for the indicated time points. Growth kinetics was measured as a function of absorbance read at 600 nm using a spectrophotometer and cultures were also plated on 7H11 medium to enumerate corresponding CFUs.
In vivo mouse model for TB and treatment with pharmacological inhibitors
BALB/c mice (n = 24) were infected with mid-log phase Mtb H37Rv, using a Madison chamber aerosol generation instrument calibrated to 200 CFU/animal. Aerosolized animals were maintained in securely commissioned BSL3 facility. Post 28 d of established infection, mice were administered eight intra-peritoneal doses of gefitinib (50 mg/kg) [Citation27] or JQ1 (6.125 mg/kg) (modified from [Citation12]) every alternate day over 16 d. On 44th day post inhibitor treatment, mice were sacrificed, spleen and left lung lobe were homogenized in sterile PBS, serially diluted and plated on 7H11 agar containing OADC to quantify CFU. Upper right lung lobes were fixed in formalin, and processed for hematoxylin and eosin staining, or immunohistochemistry, and immunofluorescence analyses.
Hematoxylin and eosin staining
Microtome sections (5 μm) were obtained from formalin-fixed, paraffin-embedded mouse lung tissue samples using Leica RM2245 microtome. Deparaffinized and rehydrated sections were subjected to hematoxylin staining followed by eosin staining as per manufacturer instructions. After dehydrating, sections were mounted using DPX (Fisher Scientific, Q18404). Sections were kept for drying overnight and handed over to consultant pathologist for blinded analyses.
Immunohistochemistry (IHC)
3.6% formaldehyde-fixed, decalcified tissues were embedded in paraffin and microtome sections (5 µm) were obtained using Leica RM2245 microtome (Leica, Germany). Deparaffinized sections were subjected to antigen retrieval by boiling in 10 mM citrate buffer (pH 6.0) for 15 min; treated with 1% H2O2 for 10 min in dark and blocked with 5% BSA in PBST for 1 h at room temperature. Post washing with PBST, the tissue sections were further incubated with primary antibodies in 5% BSA in PBST overnight. After incubation, the sections were washed with PBST and incubated with anti-rabbit HRP-conjugated secondary antibody for 90 min. The sections were stained using 0.05% diaminobenzidine in 0.03% H2O2 solution. Finally, it was counterstained with hematoxylin, dehydrated and mounted in DPX (Fisher Scientific, Q18404). Axio Scope.A1 microscope (Zeiss, Germany) was used to image the stained tissue sections at the indicated magnification. All experiments were performed with appropriate isotype-matched control antibodies.
Cryosection preparation and immunofluorescence (IF)
The fixed lung pieces were placed in the optimal cutting temperature (OCT) media (Jung, Leica, 14020108926). Cryosections of 10 µm were prepared using Leica CM 1510 S or Leica CM 3050 S cryostat and then stored at −80°C. For in vitro experiments, cells were fixed with 3.6% formaldehyde for 30 min at room temperature. Fixed samples from both in vitro and in vivo sources were blocked with 2% BSA in PBST (containing 0.02% saponin [Sigma-Aldrich, S4521]) for 1 h. After blocking, samples were stained with the indicated antibodies at 4°C overnight, followed by incubation with DyLight 488- (Thermo Fisher Scientific, 35502), Alexa Fluor 555 (Cell Signaling Technology, 4413) or Alexa Fluor 647 (BioLegend, 406414)-conjugated secondary antibodies for 2 h and nuclei were stained with DAPI. For EGFR and p-EGFR staining, following infection, cells were washed with PBS thrice and incubated with anti-EGFR or anti-p-EGFR antibody (in 2% BSA in PBS) at room temperature for 30 min. Cells were washed with PBS and incubated with anti-Rabbit Alexa-555 antibody for 30 min at room temperature. Post this, cells were washed in PBS and fixed with 3.7% PFA for 30 min, washed and stained with DAPI. All the samples were mounted on glycerol (Sigma-Aldrich, G5516) medium. Confocal images were taken with Zeiss LSM 710 Meta confocal laser scanning microscope (Carl Zeiss AG, Germany) using a plan-Apochromat 63X/1.4 Oil DIC objective (Carl Zeiss AG, Germany) and images were analyzed using ZEN 2009 software.
Lipid droplet staining
Lipid droplets were stained using neutral lipid dye BODIPY 493/503 (Invitrogen, D3922). Formaldehyde-fixed cells or tissue sections were stained with BODIPY (10 μg/ml) for 30 min at room temperature. Cells/tissues were washed, and nuclei were stained with DAPI. After washing with PBS, samples were mounted on glycerol and visualized and analyzed by confocal microscopy.
Enzyme linked immunosorbent assay (ELISA)
Cell-free lung homogenates were used for performing ELISA for VEGFA as per the manufacturer’s instructions. Briefly, 96-well flat bottom plates (Nunc MaxiSorp; Thermo Scientific, 44–2404-21) were coated with specific capture antibodies overnight at 4°C followed by three washes with PBST (PBS with 0.05% Tween 20). After blocking with 1% BSA for 1 h at room temperature, wells were washed and incubated with lung homogenates for 2 h. Post three washes with PBST, wells were incubated with respective detection antibodies for 2 h at room temperature. The wells were washed and incubated with streptavidin-HRP antibody (Peprotech, 900-K99) for 30 min at room temperature. The reactions were developed with 3,3ʹ,5,5ʹ-tetramethylbenzidine (TMB; Sigma-Aldrich, T8768) followed by arrest using 7% H2SO4 and the absorbance was measured at 450 nm using an ELISA reader (Tecan, Switzerland).
Statistical analysis
Levels of significance for comparison between samples were determined by the Student’s t-test, one-way ANOVA and two-way ANOVA followed by Bonferroni’s multiple-comparisons. The data in the graphs are expressed as the mean ± S.E. for the values from at least 3 or more independent experiments and P values < 0.05 were defined as significant. GraphPad Prism software (5.0, 8.0 and 9.0 versions, GraphPad Software, USA) was used for all the statistical analyses.
Supplemental Material
Download Zip (219 MB)Acknowledgments
We thank CAF, IISc for maintaining and providing mice for experimentation. BRD4 WT construct was provided by Dr. Rab Prinjha and Dr. Neil Garton from Glaxo Smith Kline (Deposited at GSK by Dr. Angela Bridges, BRAD Biochemistry, UK). PI3K dominant negative (PIK3R/p85 DN) construct was a kind research gift from Dr. Kumaravel Somasundaram, Department of Microbiology and Cell Biology, IISc. mRFP1-EGFP-PLIN2 construct was kindly provided by Dr. Mark A. McNiven, Mayo Clinic, Rochester, USA. We acknowledge BSL-3 facility at Center for Infectious Disease Research (CIDR) for permitting our in vitro and in vivo experiments with Mtb H37Rv. We thank Dr. Ravi Manjithaya, JNCASR, Bengaluru, India for kind suggestions.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
Related Research Data
References
- Peyron P, Vaubourgeix P, Poquet Y, et al. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog. 2008;4:e1000204.
- Daniel J, Maamar H, Deb C, et al. Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages. PLoS Pathog. 2011;7:e1002093.
- Knight M, Braverman J, Asfaha K, et al. Lipid droplet formation in Mycobacterium tuberculosis infected macrophages requires IFN-?/HIF-1a signaling and supports host defense. PLoS Pathog. 2018;14(1):e1006874.
- Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation, and immunity. Nat Rev Immunol. 2013;13(10):722–737.
- Kim JK, Kim TS, Basu J, et al. MicroRNA in innate immunity and autophagy during mycobacterial infection. Cell Microbiol. 2017;19(1):e12687.
- Holla S, Kurowska-Stolarska M, Bayry J, et al. Selective inhibition of IFNG-induced autophagy by miR155- and miR31-responsive WNT5A and SHH signaling. Autophagy. 2014;10(2):311–330.
- Kumar D, Nath L, Kamal AM, et al. Genome-wide analysis of the host intracellular network that regulates survival of Mycobacterium tuberculosis. Cell. 2010;140:731–743.
- Ghorpade DS, Holla S, Sinha AY, et al. Nitric oxide and KLF4 protein epigenetically modify class II transactivator to repress major histocompatibility complex II expression during Mycobacterium bovis bacillus Calmette-Guerin infection. J Biol Chem. 2013;288:20592–20606.
- Singh V, Prakhar P, Rajmani RS, et al. Histone methyltransferase SET8 epigenetically reprograms host immune responses to assist mycobacterial survival. J Infect Dis. 2017;216::477–488.
- Holla S, Prakhar P, Singh V, et al. MUSASHI-mediated expression of JMJD3, a H3K27me3 demethylase, is involved in foamy macrophage generation during mycobacterial infection. PLoS Pathog. 2016;12:e1005814.
- Mahadik K, Prakhar P, Rajmani RS, et al. c-Abl-TWIST1 epigenetically dysregulate inflammatory responses during mycobacterial infection by co-regulating Bone Morphogenesis Protein and miR27a. Front Immunol. 2018;9:85.
- Cheng CY, Gutierrez NM, Marzuki MB, et al. Host sirtuin 1 regulates mycobacterial immunopathogenesis and represents a therapeutic target against tuberculosis. Sci Immunol. 2017;2(9):eaaj1789.
- Belkina AC, Nikolajczyk BS, Denis GV. BET protein function is required for inflammation: brd2 genetic disruption and BET inhibitor JQ1 impair mouse macrophage inflammatory responses. J Immunol. 2013;190:3670–3678.
- McKinney CC, Kim MJ, Chen D, et al. Brd4 activates early viral transcription upon Human Papillomavirus 18 infection of primary keratinocytes. MBio. 2016;7(6):e01644–16.
- Ren K, Zhang W, Chen X, et al. An epigenetic compound library screen identifies BET inhibitors that promote HSV-1 and −2 replication by bridging P-TEFb to viral gene promoters through BRD4. PLoS Pathog. 2016;12:e1005950.
- Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell. 2014;54:728–736.
- Sakurai N, Inamochi Y, Inoue T, et al. BRD4 regulates adiponectin gene induction by recruiting the P-TEFb complex to the transcribed region of the gene. Sci Rep. 2017;7:11962.
- Sakamaki JI, Wilkinson S, Hahn M, et al. Bromodomain Protein BRD4 is a transcriptional repressor of autophagy and lysosomal function. Mol Cell. 2017;66:517–532 e519.
- Ramakrishnan L. Revisiting the role of the granuloma in tuberculosis. Nat Rev Immunol. 2012;12:352–366.
- Kim M, Wainwright HC, Locketz M, et al. Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol Med. 2010;2(7):258–274.
- Osherov N, Ben-Ami R. Modulation of host angiogenesis as a microbial survival strategy and therapeutic target. PLoS Pathog. 2016;12:e1005479.
- Zielonka TM, Demkow U, Michalowska-Mitczuk D, et al. Angiogenic activity of sera from pulmonary tuberculosis patients in relation to IL-12p40 and TNFalpha serum levels. Lung. 2011;189:351–357.
- Polena H, Boudou F, Tilleul S, et al. Mycobacterium tuberculosis exploits the formation of new blood vessels for its dissemination. Sci Rep. 2016;6:33162.
- Datta M, Via LE, Kamoun WS, et al. Anti-vascular endothelial growth factor treatment normalizes tuberculosis granuloma vasculature and improves small molecule delivery. Proc Natl Acad Sci U S A. 2015;112:1827–1832.
- Xu Y, Wang L, Zimmerman MD, et al. Matrix metalloproteinase inhibitors enhance the efficacy of frontline drugs against Mycobacterium tuberculosis. PLoS Pathog. 2018;14:e1006974.
- Kurimchak AM, Shelton C, Duncan KE, et al. Resistance to BET bromodomain inhibitors is mediated by kinome reprogramming in ovarian cancer. Cell Rep. 2016;16:1273–1286.
- Liu F, Hon GC, Villa GR, et al. EGFR mutation promotes glioblastoma through epigenome and transcription factor network remodeling. Mol Cell. 2015;60:307–318.
- Stanley SA, Barczak AK, Silvis MR, et al. Identification of host-targeted small molecules that restrict intracellular Mycobacterium tuberculosis growth. PLoS Pathog. 2014;10:e1003946.
- Wen X, Klionsky DJ. BRD4 is a newly characterized transcriptional regulator that represses autophagy and lysosomal function. Autophagy. 2017;13(11):1801–1803.
- Feng L, Hu J, Zhang W, et al. RELL1 inhibits autophagy pathway and regulates Mycobacterium tuberculosis survival in macrophages. Tuberculosis (Ednib). 2020;120:101900.
- da Motta LL, Ledaki I, Purshouse K, et al. The BET inhibitor JQ1 selectively impairs tumour response to hypoxia and downregulates CA9 and angiogenesis in triple negative breast cancer. Oncogene. 2017;36:122–132.
- Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135.
- Ding S, Qu Y, Yang S, et al. Novel miR-1958 promotes Mycobacterium tuberculosis survival in RAW 264.7 cells by inhibiting autophagy via Atg5. J Microbiol Biotechnol. 2019;29(6):989–998.
- Camare C, Pucelle M, Negre-Salvayre A, et al. Angiogenesis in the atherosclerotic plaque. Redox Biol. 2017;12:18–34.
- Alatas F, Alatas O, Metintas M, et al. Vascular endothelial growth factor levels in active pulmonary tuberculosis. Chest. 2004;125:2156–2159.
- Jaisinghani N, Dawa S, Singh K, et al. Necrosis driven triglyceride synthesis primes macrophages for inflammation during Mycobacterium tuberculosis infection. Front Immunol. 2018;9:1490.
- Wong KW, Jacobs WR Jr. Postprimary tuberculosis and macrophage necrosis: is there a big ConNECtion? MBio. 2016;7:e01589–01515.
- Offer S, Menard JA, Perez JE, et al. Extracellular lipid loading augments hypoxic paracrine signaling and promotes glioma angiogenesis and macrophage infiltration. J Exp Clin Cancer Res. 2019;38:241.
- Miyamoto S, Suzuki T, Muto S, et al. Positive and negative regulation of the cardiovascular transcription factor KLF5 by p300 and the oncogenic regulator SET through interaction and acetylation on the DNA-binding domain. Mol Cell Biol. 2003;23:8528–8541.
- Gao Y, Wu K, Chen Y, et al. Beyond proliferation: KLF5 promotes angiogenesis of bladder cancer through directly regulating VEGFA transcription. Oncotarget. 2015;6:43791–43805.
- Holbro T, Hynes NE. ErbB receptors: directing key signaling networks throughout life. Annu Rev Pharmacol Toxicol. 2004;44:195–217.
- Lindsey S, Langhans SA. Crosstalk of oncogenic signaling pathways during epithelial-mesenchymal transition. Front Oncol. 2014;4:358.
- Boro M, Singh V, Balaji KN. Mycobacterium tuberculosis-triggered Hippo pathway orchestrates CXCL1/2 expression to modulate host immune responses. Sci Rep. 2016;6:37695.
- Dai X, She P, Chi F, et al. Phosphorylation of angiomotin by Lats1/2 kinases inhibits F-actin binding, cell migration, and angiogenesis. J Biol Chem. 2013;288:34041–34051.
- Kim J, Kim YH, Park DY, et al. YAP/TAZ regulates sprouting angiogenesis and vascular barrier maturation. J Clin Invest. 2017;127:3441–3461.
- Reddy RJ, Gajadhar AS, Swenson EJ, et al. Early signaling dynamics of the epidermal growth factor receptor. Proc Natl Acad Sci U S A. 2016;113:3114–3119.
- Ouimet M, Koster S, Sakowski E, et al. Mycobacterium tuberculosis induces the miR-33 locus to reprogram autophagy and host lipid metabolism. Nat Immunol. 2016;17(6):677–686.
- Kolodgie FD, Gold HK, Burke AP, et al. Intraplaque hemorrhage and progression of coronary atheroma. N Engl J Med. 2003;349:2316–2325.
- Tough DF, Prinjha RK. Immune disease-associated variants in gene enhancers point to BET epigenetic mechanisms for therapeutic intervention. Epigenomics. 2016;9:573–584.
- de Medeiros RB. Sequential chromatin immunoprecipitation assay and analysis. Methods Mol Biol. 2011;791:225–237.
- Truax AD, Greer SF. ChIP and Re-ChIP assays: investigating interactions between regulatory proteins, histone modifications, and the DNA sequences to which they bind. Methods Mol Biol. 2012;809:175–188.