
ABSTRACT
Efficient degradation of autophagic vacuoles (AVs) generated at axon terminals by mature lysosomes enriched in the cell body represents an exceptional challenge that neurons face in maintaining cellular homeostasis. Here, we discuss our recent findings revealing a lipid-mediated impairment of lysosome transport to distal axons contributing to axonal AV accumulation in the neurodegenerative lysosomal storage disorder Niemann-Pick disease type C (NPC). Using transmission electron microscopy, we observed a striking buildup of endocytic and autophagic organelles in NPC dystrophic axons, indicating defects in the clearance of organelles destined for lysosomal degradation. We further revealed that elevated cholesterol on NPC lysosome membranes abnormally sequesters motor-adaptors of axonal lysosome delivery, resulting in impaired anterograde lysosome transport into distal axons that disrupts maturation of axonal AVs during their retrograde transport route. Together, our study demonstrates a mechanism by which altered membrane lipid composition compromises axonal lysosome trafficking and positioning and shows that lowering lysosomal lipid levels rescues lysosome transport into NPC axons, thus reducing axonal autophagic stress at early stages of NPC disease.
Lysosomes receive and degrade macromolecules from multiple membrane trafficking routes including the endocytic and autophagic pathways. Given their highly polarized morphology, neurons require efficient macroautophagic/autophagic and endolysosomal trafficking and maturation to maintain effective clearance capacities throughout the cell. In axons, endocytic and autophagic cargos move along a retrograde transport route toward the cell body, where biosynthetic and degradative compartments are relatively enriched. One intriguing feature of axonal autophagy is the observation that autophagosomes mature and become increasingly acidified as they travel from distal to proximal. This process is thought to promote encounters with endolysosomes, thereby facilitating maturation and clearance of autophagic cargo. Axonal accumulation of autophagosomes and lysosome-like organelles characterizes the early stages of several neurodegenerative diseases, highlighting the importance of axonal organelle trafficking to neuron survival. However, the impact of lysosome anterograde transport and positioning on axonal dystrophy and its contribution to the pathogenesis of neurodegenerative diseases remain largely unknown.
Niemann-Pick disease type C (NPC) is a neurodegenerative lysosomal storage disorder characterized by lipid storage in endolysosomes. One pathologic feature that occurs before symptom onset and neuron loss in NPC mice is axonal dystrophy, which consists of swellings along axons containing accumulated organelles, suggesting that defects in the trafficking of these axonal organelles contribute to early NPC pathology. However, the mechanisms underlying these pathological events in NPC neurons remain obscure. Here, we discuss our recent work uncovering a lipid-mediated impairment of axonal lysosome transport contributing to autophagosome accumulation in NPC axons [Citation1].
We first characterized the composition of organelle accumulations in dorsal root and cortical neuron axons of symptomatic NPC mice, representing two regions vulnerable to neurodegeneration in NPC. Using transmission electron microscopy, we observed a substantial buildup of endocytic and autophagic organelles in NPC dystrophic axons, which predominantly included multivesicular bodies, multilamellar vesicles, and autophagic vacuoles (AVs). Because a global failure in axonal transport would lead to more diverse organelle accumulations, these findings suggested that impaired maturation and degradation of lysosomal cargos likely underlies early axonal pathology in NPC.
Given that most axonal autophagosomes travel toward the soma for degradation, we next tested whether defects in AV retrograde transport contribute to their axonal accumulation in NPC. Consistent with a defect in AV maturation, we observed a robust increase in LC3-labeled AVs in presymptomatic NPC axons. However, unexpectedly, these axonal autophagosomes displayed similar motility patterns compared to those in control axons, moving predominantly in the retrograde direction toward the cell body. Because axonal AV maturation is linked to their retrograde transport, this was a surprising finding and suggested that other factors contribute to AV accumulation in NPC axons.
Degradation of endocytic and autophagic cargos depend on their dynamic interactions with lysosomes, which supply the acid hydrolases required for substrate degradation. Therefore, we next examined axonal lysosome distribution using LAMP1 (lysosomal associated membrane protein 1) in combination with activity-based probes that specifically label active forms of the lysosomal hydrolases GBA/glucocerebrosidase (glucosylceramidase beta) and CTSD (cathepsin D). Fluorescence microscopy analysis revealed reduced lysosome density in NPC axons but an expanded lysosomal system in the cell body, while western blot showed no reduction in the total levels of these lysosomal proteins between the genotypes. These results indicated that reduced axonal lysosome density in NPC does not result from an overall reduction in neuronal lysosome levels but suggested a change in lysosome distribution at presymptomatic stages.
This prompted us to test whether lysosomes were not being transported properly to distal axons in NPC. Using microfluidic devices that provide physical and fluidic separation of axons from neuronal cell bodies and dendrites, we monitored axonal lysosome delivery by spatially loading activity-based lysosome probes in the soma/dendritic chamber followed by live imaging distal axons in the axon chamber. Lysosomes visualized in axons following this soma-restricted labeling represent those transported from the cell body. We observed a progressive reduction in soma-labeled lysosomes in NPC axons relative to WT, indicating impaired lysosome transport into distal axons. Consistently, time-lapse imaging of live cortical neurons showed reduced anterograde lysosome transport in NPC distal axons compared with WT, reflecting a specific defect in lysosome delivery.
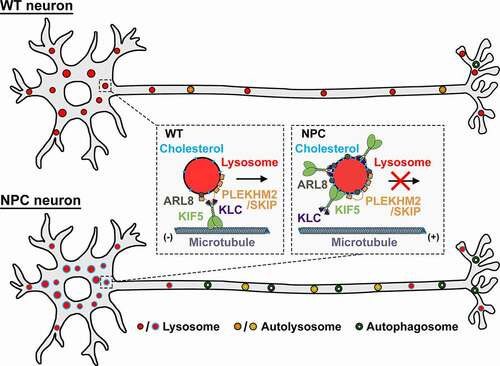
Impaired axonal lysosome delivery suggested a disruption to the ARL8-PLEKHM2/SKIP-kinesin-1 motor complex driving lysosome transport into axons. We therefore investigated recruitment of this complex to lysosomes using STED super-resolution imaging and biochemical endolysosomal isolations. Unexpectedly, we observed that ARL8 and kinesin-1, but not PLEKHM2/SKIP, are aberrantly sequestered on the surface of NPC lysosomes, reflecting accumulation of improperly assembled motor-adaptor complexes on NPC lysosome membranes. This sequestration occurs independent of PLEKHM2/SKIP, ARL8 GTPase activity, and the kinesin-1 cargo binding domain, suggesting their nonfunctional accumulation. Co-immunoprecipitation revealed reduced interaction between ARL8 and PLEKHM2/SKIP in NPC mouse brain homogenates compared with WT, further suggesting disrupted formation of the motor-adaptor-effector complexes mediating lysosome transport into axons in NPC.
The stabilization of ARL8 and kinesin-1 on NPC lysosome membranes occurred in the absence of significant changes to their total protein levels in NPC neurons, suggesting these changes were mediated locally at the lysosome membrane. Notably, we observed that NPC neuronal lysosomes display elevated membrane cholesterol levels, indicating that altered membrane lipid composition may be driving the abnormal motor-adaptor sequestration. Consistent with this notion, pharmacological reduction of lysosomal membrane cholesterol with 2-hydroxypropyl-β-cyclodextrin (HPCD) released ARL8 and kinesin-1 sequestration, thereby rescuing axonal lysosome delivery by reforming functional motor complexes, and reducing autophagosome accumulation in presymptomatic NPC neurons.
Taken together, our study supports a model in which alterations in lipid composition within NPC lysosome membranes impairs their axonal trafficking and positioning, contributing to autophagosome accumulation in NPC axons (). These findings highlight the role of soma-derived axonal lysosomes in maintaining axonal integrity and demonstrate that impaired lysosome transport to distal axons disrupts maturation and progression of the autophagy pathway and contributes to altered axonal homeostasis in NPC neurons.
Figure 1. Schematic model showing lipid-mediated sequestration of motor-adaptor proteins impairs axonal lysosome delivery, contributing to autophagic stress in NPC axons. In WT neurons, the ARL8-PLEKHM2/SKIP-kinesin-1 complex is appropriately recruited to and assembled on lysosome membranes in the soma, driving lysosome transport into axons to facilitate axonal autophagosome maturation and clearance. In NPC neurons, altered membrane lipid composition on somatic lysosomes sequesters ARL8 and kinesin-1 independent of PLEKHM2/SKIP, disrupting lysosome transport to distal axons. Inefficient lysosome delivery impairs axonal autophagosome maturation, resulting in increased autophagic stress in presymptomatic NPC axons. Reducing lysosomal membrane cholesterol with HPCD treatment releases ARL8 and kinesin-1 sequestration, thus rescuing lysosome transport into axons and reducing axonal autophagosome accumulation at early stages of NPC disease
Disclosure Statement
The authors declare no competing interests.
Additional information
Funding
Reference
- Roney JC, Li S, Farfel-Becker T, et al. Lipid-mediated motor-adaptor sequestration impairs axonal lysosome delivery leading to autophagic stress and dystrophy in Niemann-Pick type C. Dev Cell. 2021 Apr 13;56(10):1452–1468.e8.