
ABSTRACT
Human genetics and loss-of-function studies revealed a critical role for macroautophagy/autophagy in host defense. The autophagic delivery of intracellular pathogens to lysosomes is a central mechanism of innate immunity; thus, augmentation of host xenophagy represents a promising and powerful approach to combat infections. The precise mechanisms required for autophagosome biogenesis and maturation, however, remain unclear. Using a targeted genetic screen against phosphoinositide kinases and phosphatases, our recent work identified an essential role for the phosphoinositide phosphatase SACM1L/SAC1 in xenophagy. Re-expression of wild-type or catalytically-dead SACM1L in CRISPR knockout cells confirmed that SACM1L enzymatic activity is required to suppress replication of intracellular Salmonella. Time-dependent, quantitative and live confocal imaging demonstrated that SACM1L-deficient cells accumulate phosphatidylinositol-4-phosphate (PtdIns4P) on bacteria-containing autophagosomes, resulting in delayed fusion with degradative lysosomes and reduced bacterial killing. We further discovered that the secreted Salmonella effector protein SteA, which specifically binds PtdIns4P, exacerbates the SACM1L-dependent delay in autophagosomal maturation. These findings reveal a relationship in which the balance between host defense and bacterial survival depends upon autophagosomal membrane composition.
Autophagosomal membranes are composed predominantly of phospholipids including phosphatidylinositols, which are modified into seven distinct phosphorylated forms that identify membrane compartments and signal through protein interactions. We interrogated the role of lipid membrane composition and signaling in xenophagy regulation by examining the effects of small interfering RNA (siRNA) directed against phosphoinositide kinases and phosphatases on intracellular Salmonella replication, and uncovered SACM1L/SAC1, a transmembrane PtdIns4P phosphatase, as an essential regulator of xenophagy. This finding initiated our investigation into how loss of SACM1L disrupts this conserved, multi-step host defense mechanism [Citation1].
Our initial observations suggested the importance of PtdIns4P regulation in restricting bacterial replication, as siRNAdirected against SACM1L and the PtdIns4P kinase PI4K2A/PI4K2α had opposing phenotypes. Generating SACM1L CRISPR knockout cells and re-expressing wild-type or catalytically-dead protein confirmed that SACM1L phosphatase activity is required to suppress bacterial replication. As Salmonella is a known target of xenophagy, we speculated that in the absence of SACM1L cells accumulate PtdIns4P on membranes, which disrupts signaling events essential for bacteria clearance. Considering core protein complexes that function in autophagosomal biogenesis are well conserved between nonselective and selective autophagy processes, we anticipated that loss of SACM1L would reduce autophagic flux or clearance of cargo. Surprisingly, we did not detect defects in nonselective nor other forms of selective autophagy, including PRKN/parkin-dependent mitophagy and aggrephagy, indicating that SACM1L functions specifically in xenophagy (). These findings advanced our understanding of the importance of membrane composition in mechanisms regulating autophagy processes.
Figure 1. Function of SACM1L PtdIns4P phosphatase in autophagy. Loss of SACM1L leads to the accumulation of PtdIns4P on cellular membranes, including autophagosomes, and a defect in xenophagy-dependent restriction of intracellular bacterial replication. Fusion of lysosomes with nonselective autophagosomes, as well as those induced through selective mitophagy and aggrephagy, are unaffected by loss of SACM1L expression. Salmonella-secreted effector and PtdIns4Pbinding protein SteA contributes to the xenophagy defect by blocking lysosomal fusion, and thus, shifting the balance in favor of bacterial replication. Loss of both SACM1L and Salmonella SteA restores the host defense mechanism.
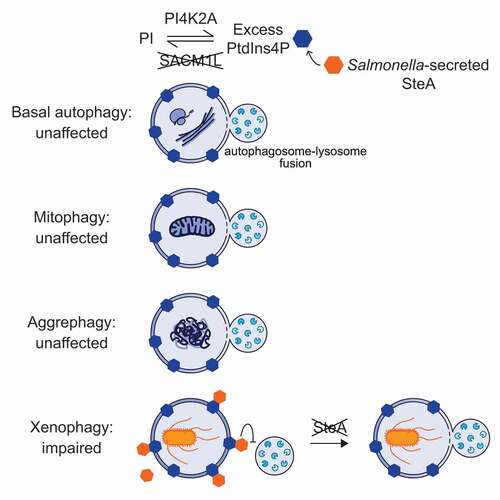
Autophagy processes are highly controlled by spatiotemporal regulation of core protein complexes; therefore, we used time-dependent and live confocal microscopy to dissect the steps of xenophagy and understand the role of SACM1L. In the initial stages of selective autophagy, cargo receptors target specific cytoplasmic content to nascent phagophores by connecting ubiquitinated substrates with LC3 on phagophore membranes. Ubiquitinated Salmonella are predominantly targeted by two cargo receptors, CALCOCO2/NDP52 and SQSTM1/P62. Whereas our quantitative image analysis did not identify a SACM1L-dependent defect in ubiquitination or targeting of bacteria, it did reveal a higher percentage of bacteria associated with both CALCOCO2 and SQSTM1 as well as membrane-bound LC3 at time points after infection when detection of these proteins should decline due to degradation and recycling, as observed in wild-type cells. These data were the first indication that loss of SACM1L may delay maturation of bacteria-containing autophagosomes.
Following recognition and targeting, phagophore membranes expand and encapsulate bacteria to form autophagosomes prior to fusion with lysosomes to complete the degradative process. Our data demonstrated that loss of SACM1L impairs lysosomal fusion with bacteria-containing autophagosomes. In a host cell, however, bacterial pathogens have several possible fates. Intracellular Salmonella, for example, can reside and replicate within Salmonella-containing vacuoles (SCVs), escape the SCV and replicate in the cytosol, or be targeted and cleared by xenophagy. Using live confocal microscopy, we imaged intracellular Salmonella over time and observed a population of bacteria in SACM1L-deficient cells that are targeted by autophagy machinery but do not accumulate lysosomal markers, in contrast to wild-type cells. Interestingly, autophagy markers became undetectable at later time points, and the bacteria began to rapidly replicate in the host cytosol. These findings were consistent with our initial bacterial replication phenotype, as bacteria targeted but not degraded by lysosomal enzymes continue to replicate. The xenophagy-specific role for SACM1L-dependent PtdIns4P regulation, however, remained unclear.
Knowing that intracellular pathogens evolve strategies to subvert host defense mechanisms, we examined the role of SteA, a Salmonella-secreted effector and PtdIns4P-binding protein, on the SACM1L-dependent xenophagy defect. We speculated that if SteA bound to PtdIns4P on bacteria-containing autophagosomes inhibits lysosomal fusion, then SACM1L-deficient cells would restrict replication of ΔsteA Salmonella. Our data confirmed that both wild-type and SACM1L-deficient cells reduce replication of Salmonella lacking SteA or expressing a mutant SteA unable to bind PtdIns4P. Importantly, replication of ΔsteA Salmonella in wild-type cells is significantly reduced relative to that of wild-type Salmonella, indicating that SteA also promotes bacterial replication in the presence of SACM1L. These data support the model that during xenophagy SACM1L and PI4K2A regulate PtdIns4P levels on autophagosomes for proper membrane signaling and trafficking, while Salmonella secrete SteA, which in part binds PtdIns4P on bacteria-containing autophagosomes to promote bacterial survival. In the event of SACM1L loss, PtdIns4P accumulates on autophagosomal membranes and enhances the effect of SteA, blocking lysosomal fusion and increasing bacterial replication.
Collectively, our study is an important example of the delicate balance between innate host defense restricting bacterial replication and bacterial factors promoting intracellular survival. We revealed the significance of SACM1L-dependent PtdIns4P regulation on xenophagy-specific autolysosomal maturation and uncovered a new role for Salmonella SteA in bacterial pathogenesis. Such identification of host mechanisms that prevent infection is a compelling area of research with the potential to inform new treatment strategies.
Disclosure statement
R.J.X. is co-founder of Jnana Therapeutics and Celsius Therapeutics. These organizations had no roles in this study.
Additional information
Funding
Reference
- Liu K, Kong L, Graham DB, et al. SAC1 regulates autophagosomal phosphatidylinositol-4-phosphate for xenophagy-directed bacterial clearance. Cell Rep. 2021 Jul 27;36(4):109434.