
ABSTRACT
ULK1 kinase is the gatekeeper of canonical macroautophagy (hereafter referred to as autophagy) phosphorylating an array of substrates critical for autophagosome biogenesis. To uncover if ULK1 has broader functions also regulating subsequent steps of autophagosome turnover, i.e., maturation, lysosomal fusion, and degradation, we performed a set of unbiased phosphoproteomic experiments employing mouse and human cells in combination with genetic and environmental perturbations. We characterized more than 1,000 potential ULK1 target sites of which many affect proteins known to be involved in all phases of the autophagosome life cycle. To better understand which of these 1,000 phosphosites were directly phosphorylated by ULK1, in contrast to downstream kinases being activated or phosphatases being inhibited by ULK1, we developed a proteome-scale in vitro kinase assay and characterized 187 phosphosites on 157 proteins as bona fide ULK1 target sites. Interestingly, our results highlight an intricate crosstalk between ULK1 and protein phosphatases. Focusing on STRN (striatin), a regulatory subunit of PPP2/PP2A (protein phosphatase 2), we identified a positive feedback loop linked to ULK1 and promoting autophagy.
Autophagy is a well-known lysosomal degradation pathway in eukaryotic cells and commonly acts in a cytoprotective manner. This process can function in a constitutive, nonselective as well as in a selective fashion as part of cellular stress responses. Autophagy induction is regulated by phosphorylation-based signaling events. The major autophagy activating kinase complexes are the protein kinase complex containing ULK1 (or its homolog ULK2) and its target, the class III phosphatidylinositol 3-lipid kinase complex containing PIK3C3/VPS34. Both are critical for de novo formation of double membrane vesicles, so called autophagosomes, which are a hallmark of autophagy. In mammalian cells, the major autophagy inhibitory kinase complex is MTORC1, which stimulates anabolic reactions and cell growth and inhibits autophagy by phosphorylating among others ULK1 itself and its complex member ATG13. For ULK1 activation, i.e., autophagy induction, these inhibitory MTORC1 phosphorylations on Ser638 and Ser758 of human ULK1 have to be removed by protein phosphatases. Thus, it was shown that depending on the autophagy inducing conditions, PPP2 and PPP1 complexes are critical for the removal of these inhibitory modifications on MTORC1 phosphorylation sites, leading to the full activation of ULK1 kinase and with this to functional autophagy. PPP2 protein phosphatases form multimeric holo-complexes that are trimeric protein assemblies made up of a catalytic, scaffold, and regulatory subunit. Due to the combination of several distinct gene products and isoforms several tens of distinct holo-complexes are thought to exist in a given cell type and under specific conditions. It is entirely unclear how these complexes are regulated in autophagy and how different complexes affect autophagy.
In our recent publication [Citation1], we performed in vivo phosphoproteomic experiments employing ulk1−/− ulk2−/− double-knockout (DKO) mouse embryonic fibroblasts expressing human ULK1 as well as A549 adenocarcinomic human alveolar cells and kept them in starvation medium or treated them with rapamycin to inhibit MTORC1 and to induce autophagy. To identify direct ULK1 targets on a global scale, we developed a proteome wide on-bead in vitro kinase assay (OBIKA) under native conditions. Native protein complexes were immobilized on NHS-activated sepharose beads, followed by lambda phosphatase treatment to remove endogenous phosphorylations and by inhibition of endogenous kinases using a covalent kinase inhibitor. Subsequent in vitro kinase assays were set up using purified wildtype ULK1 and kinase dead ULK1 as a negative control. Combined analysis of in vivo and in vitro datasets identified 10,687 common sites of which a total of 1,927 sites are significantly regulated, the overlap being 187 bona fide ULK1 target sites on 157 proteins, the majority of them exhibiting the well-known ULK1 kinase motif. Many proteins known to be relevant for autophagy were identified as ULK1 targets, such as exocyst complex components, VAPA, VAPB, and HUWE1, which is crucial for mitophagy.
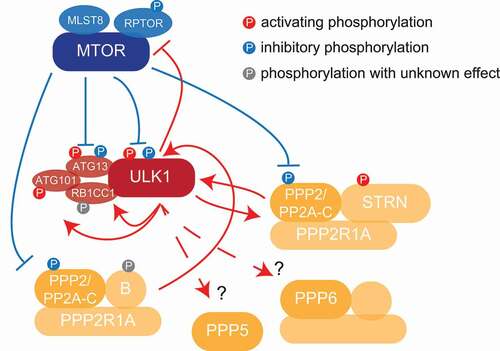
Interestingly, starvation and rapamycin treatment lead also to a significant downregulation of phosphorylation events compared to non-treated cells, indicating increased phosphatase activity under autophagy conditions. Indeed, using a calorimetric assay, we could show that overall phosphatase enzymatic activity is increased by 30% in starvation compared to growth conditions. In agreement, a network of protein phosphatases was identified as ULK1 targets, among others these included regulatory subunits of PPP6 and PPP2 holo-complexes, as well as PPP5 interaction partners ().
Figure 1. Kinase-phosphatase crosstalk in autophagy regulation. ULK1 and PPP2 are linked via a positive feedback mechanism, ULK1 activating PPP2 by phosphorylation of STRN, which in turn removes inhibitory MTORC1 sites on ULK1 itself. How ULK1 affects additional phosphatases like PPP5 and PPP6, and if this has an influence on autophagy regulation is currently not known.
We studied the ULK1 phosphorylation of STRN (striatin), a regulatory subunit of PPP2 and founding member of the evolutionarily conserved STRN-interacting phosphatase and kinase (STRIPAK) complexes, which link PPP2 complexes with germinal center kinases. Using chemical proteomic experiments assessing phosphatase activity, we enriched catalytic, scaffolding and regulatory subunits of PPP2 and PPP6 complexes in starvation- and rapamycin-treated cells. Importantly, all three members of the STRN protein family, STRN, STRN3 and STRN4, are enriched, two of them also carrying regulated phosphosites within ULK1 consensus motifs. To study a potential effect of STRN on ULK1 activity and autophagy flux, we used ATG14 Ser29 phosphorylation, LC3-II lipidation and SQSTM1/p62 puncta formation as phenotypic readouts in the presence and absence of lysosomal inhibitors. Overexpression of STRN promotes the activity of ULK1 and autophagy by removing the inhibitory MTORC1 sites p-Ser638 and p-Ser758 on ULK1, whereas shRNA-based knockdown has the opposite effect. Finally, we tested the effects of ULK1 phosphorylation of STRN Ser227 using respective site mutants. Phosphomimetic STRNS227D supports ULK1 activity and with this autophagy similar to the wild type form of STRN, whereas non-phosphorylatable STRNS227A has the opposite effect.
Altogether, we could show that PPP2 and ULK1 are linked by positive feedback, ULK1 phosphorylating the regulatory PPP2 subunit STRN, which in turn supports PPP2 activation, ULK1 dephosphorylation and increased protein degradation by autophagy. Importantly, ULK1 appears to regulate a network of phosphatases (), indicating complex kinase-phosphatase interactions, whose influence on autophagy regulation we are only beginning to understand.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Reference
- Hu Z, Sankar DS, Vu B, et al. ULK1 phosphorylation of striatin activates protein phosphatase 2A and autophagy. Cell Rep. 2021 Sep 28;36(13):109762.