
ABSTRACT
The removal of mitochondria in a programmed or stress-induced manner is essential for maintaining cellular homeostasis. To date, much research has focused upon stress-induced mitophagy that is largely regulated by the E3 ligase PRKN, with limited insight into the mechanisms regulating basal “housekeeping” mitophagy levels in different model organisms. Using iron chelation as an inducer of PRKN-independent mitophagy, we recently screened an siRNA library of lipid-binding proteins and determined that two kinases, GAK and PRKCD, act as positive regulators of PRKN-independent mitophagy. We demonstrate that PRKCD is localized to mitochondria and regulates recruitment of ULK1-ATG13 upon induction of mitophagy. GAK activity, by contrast, modifies the mitochondrial network and lysosomal morphology that compromise efficient transport of mitochondria for degradation. Impairment of either kinase in vivo blocks basal mitophagy, demonstrating the biological relevance of our findings.
Abbreviations: CCCP: carbonyl cyanide-m-chlorophenyl hydrazone; DFP: deferiprone; GAK: cyclin G associated kinase; HIF1A: hypoxia inducible factor 1 subunit alpha; PRKC/PKC: protein kinase C; PRKCD: protein kinase C delta; PRKN: parkin RBR E3 ubiquitin protein ligase.
Damaged or aged mitochondria are constantly being removed and replenished to maintain an active healthy pool of mitochondria. Mitochondria can also be removed in a programmed manner, such as in the absence of oxygen when they are no longer needed for the generation of ATP by oxidative phosphorylation. The cell has developed several mechanisms for mitochondrial quality control, including detection, disposal, and recycling of superfluous or nonfunctional parts of or whole mitochondria by autophagy, commonly referred to as mitophagy.
Mitophagy can be induced through multiple triggers and pathways. The rapid depolarization of the mitochondrial membrane potential indicates loss of mitochondrial integrity and leads to recruitment of PRKN (parkin RBR E3 ubiquitin protein ligase) and other components to clear mitochondria in a ubiquitin-dependent manner. On the contrary, environmental cues such as hypoxia or iron depletion can trigger mitochondrial turnover in a PRKN-independent manner that appears to be dependent upon HIF1A (hypoxia inducible factor 1 subunit alpha) and its response genes. Interestingly, “housekeeping” levels of mitophagy do not require PRKN, suggesting that the majority of basal mitophagy is regulated in a PRKN-independent manner.
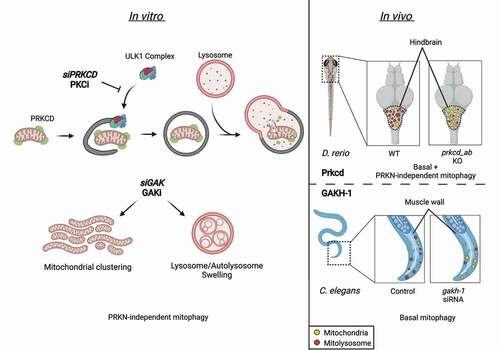
We aimed to identify new regulators of PRKN-independent mitophagy in the hope that this may highlight regulators of basal mitophagy in vivo, using the iron chelator deferiprone (DFP) that mimics a HIF1A response. Given the membrane dependence of the autophagy pathway, we focused specifically on proteins that contain domains known to interact with lipids (e.g., FYVE, PX, C1, C2 domains, etc.). Carrying out an image-based siRNA screen in U2OS cells expressing a mitochondrial matrix localized EGFP-mCherry reporter, we determined that the kinases GAK (cyclin G associated kinase) and PRKCD (protein kinase C delta) act as positive regulators of DFP-induced mitophagy [Citation1]. We validated their effect on mitophagy through several means, including biochemical activity assays, Western blot, and proteomic-based approaches. GAK and PRKC/PKC (protein kinase C) kinase inhibitors recapitulate the effects of targeted siRNA treatment, showing that their kinase activities are key for their roles as positive regulators of PRKN-independent mitophagy. In contrast, the same kinase inhibitors do not affect depolarization-induced (using carbonyl cyanide-m-chlorophenyl hydrazone [CCCP]) PRKN-dependent mitophagy in cells with PRKN-overexpression or serum and amino acid starvation (using Earle’s balanced salt solution/EBSS)-induced autophagy. Thus, GAK and PRKCD are specific to PRKN-independent mitophagy with no general effect on autophagy.
We discovered that PRKCD localizes to mitochondria irrespective of its lipid-binding domains and that it becomes degraded upon DFP- or CCCP-mediated mitophagy, indicating it remains localized to mitochondria during mitophagy. By analysis of the early autophagy markers ULK1 (unc-51 like autophagy activating kinase 1), ATG13 (autophagy related 13), and WIPI2 (WD repeat domain, phosphoinositide interacting 2), we found that PRKC inhibitors strongly prevent the formation of initiation sites during DFP treatment, likely explaining the observed inhibition of mitophagy.
GAK does not localize to mitochondria and its kinase inhibition results in more profound mitochondrial morphology changes, with mitochondrial networks clumping together into parallel stagnant networked layers. Co-treatment with CCCP causes mitochondrial fragmentation, indicating functional mitochondrial fission machinery. GAK inhibition also induces phosphorylation changes in glycolytic proteins that may indicate metabolic pathway changes. In addition, large autolysosome-like structures with multiple membrane layers are observed by electron microscopy, indicating that GAK affects lysosomal morphology. However, acidic LysoTracker-positive compartments that retain degradative ability are still observed in cells treated with the GAK inhibitor during CCCP or nutrient-starvation treatments, indicating that GAK activity is specifically required for DFP-induced mitophagy.
Having observed a role for PRKCD and GAK in PRKN-independent mitophagy at a cellular level, we wanted to examine whether either kinase was important for mitophagy on a whole organism level. GAK is an essential gene due to the presence of a C-terminal J-domain that mediates clathrin uncoating. C. elegans offered a unique opportunity to test a possible in vivo function of GAK due to the presence of two homologs to GAK, GAKH-1 that contains the kinase domain, and DNJ-25 that contains the essential J-domain. Selective silencing of gakh-1 results in inhibition of basal mitophagy in the muscle cell wall, confirming that the kinase activity is key to mediating GAK’s effect on mitophagy. This finding is of additional interest given that GAK mutations are also a risk factor for the development of Parkinson disease.
PRKCD has two homologs in zebrafish (prkcda and prkcdb) that are expressed across neuronal compartments. Using an antibody recognizing zebrafish Prkcda and Prkcdb, we identified an enriched expression in the hindbrain region of zebrafish larvae. Examination of mitophagy in this region showed that both basal and Hif1a-dependent mitophagy pathways are inhibited by CRISPR-Cas9-mediated knockout of both prkcda and prkcdb. Furthermore, double-knockout larvae display anxiety-dependent locomotory defects that may indicate neuronal abnormalities that require further investigation.
This work identifies two kinases as playing distinct roles in PRKN-independent mitophagy and offers exciting insights into the regulation of basal mitophagy in vivo (). Further work is now required to fully understand mechanistically how each kinase orchestrates its role in mitophagy and identify downstream phosphorylated substrates. This will potentially enable the specific targeting and regulation of PRKN-independent mitophagy, independently of other cellular autophagy pathways.
Figure 1. The regulation of PRKN-independent mitophagy by GAK and PRKCD. Several subtypes of autophagy and mitophagy pathways exist, including PRKN-independent mitophagy (DFP-induced), PRKN-dependent mitophagy (CCCP-induced), and starvation-induced autophagy (EBSS-induced). PRKCD localizes to mitochondria independent of its C1 or C2 domains and becomes degraded upon induction of mitophagy pathways. Recruitment of ATG13-ULK1-RB1CC1/FIP200-ATG101 to phagophore initiation sites upon induction of PRKN-independent mitophagy is blocked by PRKCD siRNA (siPRKCD) or PRKC kinase inhibitors (PRKCi). Treatment with GAK siRNA (siGAK) or GAK kinase inhibitor (GAKi) causes mitochondrial clustering and an accumulation of swollen multi-lamellar lysosomes. In vivo examination of the hindbrain region of zebrafish (D. rerio) demonstrates basal mitophagy, which is reduced by dual knockout of prkcda and prkcdb (homologs of PRKCD). Similarly, RNAi-mediated depletion of gakh-1 (GAK homolog) in worms (C. elegans) reduces basal mitophagy levels in muscle cell walls.
Disclosure statement
M.J.M. is now an employee of AstraZeneca. The remaining authors declare no competing interests.
Additional information
Funding
Reference
- Munson MJ, Mathai BJ, Ng MYW, et al. GAK and PRKCD are positive regulators of PRKN-independent mitophagy. Nat Commun. 2021;12:6101.