
ABSTRACT
Senecavirus A (SVA), an important emerging porcine virus, has outbreaks in different regions and countries each year, becoming a virus with global prevalence. SVA infection has been reported to induce macroautophagy/autophagy; however, the molecular mechanisms of autophagy induction and the effect of SVA on autophagy remain unknown. This study showed that SVA infection induced the autophagy process in the early stage of SVA infection, and the rapamycin-induced autophagy inhibited SVA replication by degrading virus 3 C protein. To counteract this, SVA utilized 2AB protein inhibiting the autophagy process from promoting viral replication in the late stage of SVA infection. Further study showed that SVA 2AB protein interacted with MARCHF8/MARCH8 and LC3 to degrade the latter and inhibit the autophagy process. In addition, we found that MARCHF8 was a positive regulator of type I IFN (IFN-I) signaling. During the autophagy process, the SVA 2AB protein targeted MARCHF8 and MAVS forming a large complex for degradation to deactivate IFN-I signaling. Together, our study reveals the molecular mechanisms of selective autophagy in the host against viruses and reveals potential viral strategies to evade the autophagic process and IFN-I signaling for successful pathogenesis.
Abbreviations: Baf A1: bafilomycin A1; Co-IP: co-immunoprecipitation; CQ: chloroquine; DAPI: 4′,6-diamidino-2-phenylindole; hpi: hours post-infection; IFN: interferon; ISG: IFN-stimulated gene; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MARCHF8/MARCH8: membrane associated ring-CH-type finger 8; MAVS: mitochondrial antiviral signaling protein; MOI: multiplicity of infection; Rapa: rapamycin; RT: room temperature; siRNA: small interfering RNA; SVA: Senecavirus A; TCID50: 50% tissue culture infectious doses.
Introduction
Senecavirus A (SVA), a non-enveloped single-stranded positive-sense RNA virus, belongs to the genus Senecavirus in the family Picornaviridae [Citation1]. SVA infection can cause severe vesicular and/or ulcerative lesions on the oral mucosa, snout, coronary bands, and hooves, and indistinguishable from the clinical symptoms caused by foot-and-mouth disease virus/FMDV, vesicular stomatitis virus/VSV, swine vesicular disease virus/SVDV and vesicular exanthema of swine virus/VESV [Citation2–6]. SVA was first detected in the United States (US) in 2002 as a cell culture contaminant, and subsequently, SVA was detected in pigs with vesicular lesions in many countries, such as Canada, US, Thailand, Brazil, and China, and caused some economic consequences to the swine industry worldwide [Citation7–11]. Therefore, more attention needs to be paid to surveillance the spread of SVA.
The SVA genome is approximately 7.3 kb and encodes a single polyprotein, which consists of the leader (L) and three major protein regions (P1, P2, and P3), and this polyprotein is subsequently processed into the structural and nonstructural proteins by proteases 2A and 3 C [Citation12,Citation13]. Although the innate immune response is the first line of host defense against viral invasion, many viruses have evolved various strategies to evade host defenses. SVA, an emerging novel picornavirus, could target and cleavage the MAVS, TRIM69/TRIF, and TANK adaptors at specific sites based on its 3 C protease (3 Cpro) properties to antagonize RLR-mediated IFN production [Citation14]. The SVA 3 Cpro reduces DDX58/RIG-I, IRF3, and IRF7 expression to block the transcription of IFNB/IFN-β, IFNA1/IFN-α1, IFNA4/IFN-α4, and IFIT2/ISG54 [Citation15,Citation16]. The SVA 3 Cpro also cleaves PABPC1 (poly(A) binding protein cytoplasmic 1) to promote viral replication [Citation17]. Therefore, the relationship between virus and host should be investigated to uncover the mechanisms of SVA replication in the infected cells.
Virus infection results in various cellular stress responses by influencing the mRNA translation, protein localization, and degradation. Macroautophagy/autophagy, one of the stress responses, degrades the damaged organelles and misfolded cytoplasmic proteins to maintain cellular homeostasis [Citation18,Citation19]. Autophagy can be activated by various viral infections and plays an important role in pathogen infection [Citation20,Citation21]. In porcine epidemic diarrhea virus (PEDV) infection, autophagy successfully induced and restricted intracellular pathogen replication by degrading the viral N protein [Citation22]. Conversely, herpes simplex virus type 1 (HSV-1) can inhibit autophagy by the HSV-1-encoded protein direct interaction with BECN1 and EIF2AK2/PKR to facilitate virus replication [Citation23,Citation24]. Nevertheless, several viruses such as influenza A virus, dengue virus, and classical swine fever virus have evolved different strategies to use autophagic vesicles for replication [Citation25–27]. It has been demonstrated that autophagy inhibited SVA replication in human cultured cells and promoted SVA replication in pig cells [Citation28,Citation29]. Therefore, the effects of autophagy on SVA replication may differ in different cells.
This study showed that autophagy was induced or inhibited by SVA infection in a time-dependent manner. Autophagy induced in the early stage of SVA infection inhibited viral replication by degrading the SVA 3 C protein. During late SVA infection, autophagy was inhibited through SVA 2AB protein-recruited MARCHF8 to ubiquitinate and degrade LC3. We also found that MARCHF8 was a positive regulator of type I IFN signaling. Upon SVA infection, 2AB protein could promote MARCHF8 and MAVS degradation to inhibit IFN-I signaling from facilitating viral replication. Our findings demonstrate the mechanism of autophagy against viral replication and reveal potential viral strategies to evade autophagy and the IFN signaling pathway for successful pathogenesis.
Results
Autophagy inhibited SVA replication by degrading the viral 3 C protein in the early stage of viral infection
Autophagy plays an important role in viral infection. To explore whether autophagy was induced by SVA infection, the levels of MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3), a marker protein of autophagy [Citation22], were examined in SVA-infected porcine kidney 15 (PK-15) cells. The results indicated that the conversion level of LC3-I to LC3-II gradually increased at 3 and 6 h post-infection (hpi) in SVA-infected PK-15 cells, as compared with mock cells (), which is consistent as previously described [Citation28,Citation29]. The induction of autophagy promoted SVA replication in SK6 cells and ST cells but inhibited SVA replication in H1299 cells and 293 T cells [Citation28], so the effect of autophagy on SVA infection might be species-specific. To determine whether autophagy is involved in the regulation of SVA replication in natural host cells. PK-15 cells were treated with rapamycin (Rapa), an inducer of autophagy, to evaluate SVA replication. As shown in , C, and D, Rapa significantly upregulated LC3-II expression, and viral protein 3D and viral RNA significantly decreased in Rapa-treated PK-15 cells. Consistent with the results, inhibition of autophagy by chloroquine (CQ), an inhibitor of autophagy, also increased SVA production (, F, and G). Compared with control, the treatment (Rapa and CQ) groups showed no differences in cell viability (data not shown). These results show that autophagy was induced by SVA infection and inhibited SVA replication.
Figure 1. SVA-induced autophagy inhibits viral replication by degrading the SVA 3 C protein in the early stage of viral infection. (A) PK-15 cells were mock-infected or infected with SVA at an MOI (multiplicity of infection) of 1 and harvested at 0, 3, and 6 hpi. The expression of SVA 3D and LC3 proteins were analyzed by Western blotting. ACTB/β-Actin was used as the sample loading control. (B, C, and D) PK-15 cells were incubated or mock incubated with Rapa for 4 h and then infected with SVA for the indicated times. The cells lysates and culture supernatants were collected to analyze SVA 3D expression and viral titers with Western blotting, real-time PCR, and TCID50 assay. (E, F, and G) PK-15 cells were incubated with CQ and then infected with SVA. The cells lysates and culture supernatants were collected to analyze SVA 3D expression and viral titers with Western blotting, real-time PCR, and TCID50 assay. (H) 293 T cells were transfected with empty vectors or plasmids encoding Flag-tagged 2AB, 2 C, 3AB, 3 C, and 3D proteins for 12 h and then treated with Rapa (6.25 μM) for 12 h. The cell lysates were analyzed by Western blotting. (I) 293 T cells were transfected with the vector expressing Flag-3 C and treated with increasing concentrations of Rapa (wedge) for 12 h. The protein level of 3 C was analyzed with Western blotting. Data are means ± SD of triplicate samples. *P < 0.05, **P < 0.01, ***P < 0.001 (two-tailed Student’s t-test).
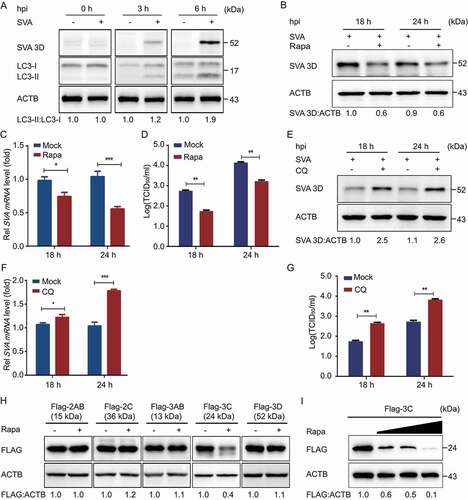
Because autophagy is an important cellular catabolic process to limit viral spread through degrading viral components [Citation22,Citation30], we hypothesized that autophagy might inhibit SVA replication by degrading viral proteins. The plasmids encoding viral proteins were constructed and transfected into cells. When Rapa induced autophagy, SVA 3 C protein was decreased in 293 T cells, but other viral proteins did not (). Furthermore, we observed that 3 C protein amount negatively correlated with the concentration of Rapa (). Altogether, these results indicated that autophagy inhibited SVA replication by degradation of viral 3 C protein.
SVA 2AB protein inhibited autophagy through degradation of LC3 protein
Although SVA induced autophagy during early infection, we hypothesize that SVA might evade autophagy for its survival. As expected, the autophagy pathway was inhibited at the late stage of SVA infection (after 12 h) (). To investigate the underlying mechanism that SVA evaded the autophagy pathway, we transfected the plasmids encoding viral proteins into 293 T cells and found that SVA 2AB protein significantly decreased LC3 protein (). Furthermore, 2AB downregulated endogenous LC3 in a dose-dependent manner (). To exclude the possibility that the downregulation of LC3 protein was caused by lower transcription of the LC3 gene, we used real-time PCR to analyze cells transfected with 2AB and found that LC3 mRNA did not alter with increased expression of 2AB (data not shown), suggesting that 2AB affected LC3 stability.
Figure 2. Autophagy is inhibited by SVA 2AB protein in the late stage of viral infection. (A) PK-15 cells were mock-infected or infected with SVA at an MOI (multiplicity of infection) of 1 and harvested at the indicated times. The expression of SVA 3D and LC3 proteins were analyzed by Western blotting. ACTB was used as the sample loading control. (B) 293 T cells were transfected with empty vectors or plasmids encoding Flag-tagged viral proteins for 24 h. The cell lysates were analyzed by Western blotting. (C) 293 T cells were transfected with the increasing concentrations of a vector expressing Flag-2AB for 24 h, and the protein was harvested for Western blotting. (D) 293 T cells were transfected with plasmids encoding Flag-2AB. Twenty-four hours later, the cells were treated with Baf A1 (0.1 μM), CQ (10 μM), Rapa (6.25 μM), or MG132 (5 μM). The cell lysates were then analyzed by Western blotting. Data are means ± SD of triplicate samples. *P < 0.05, **P < 0.01, ***P < 0.001 (two-tailed Student’s t-test).
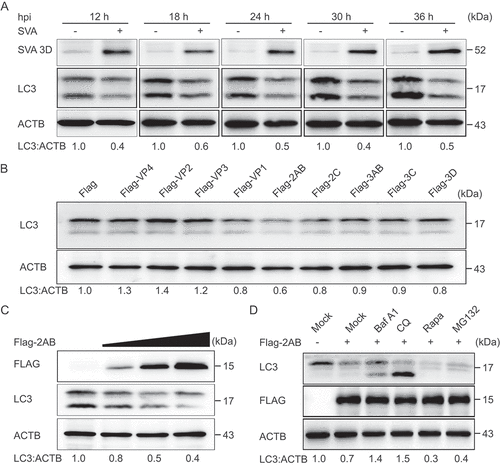
There are two major intracellular protein degradation pathways in eukaryotic cells: ubiquitin-proteasome system and autolysosome pathway [Citation31]. To identify which degradation system dominantly mediates the degradation of LC3 by 2AB protein, we treated the cells with the protein degradation pathway inhibitors and found that the degradation of LC3 mediated by 2AB could be blocked by the autophagy inhibitor CQ, bafilomycin A1 (Baf A1), but not the proteasome inhibitor MG132 (), which suggested that 2AB promoted autophagic degradation of LC3. Moreover, 2AB significantly enhanced the degradation of LC3 in Rapa-induced autophagy activation (). Collectively, these results suggested that 2AB promotes the degradation of LC3 protein through autophagy.
SVA 2AB promotes LC3 protein degradation through MARCHF8 to ubiquitinate LC3
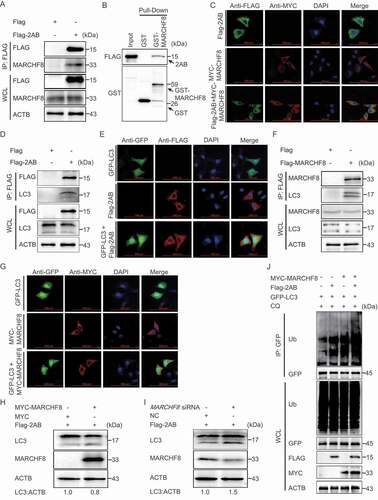
Increasing evidence suggested that autophagy is a selective process during viral infection. During selective autophagy, cargo receptors can recognize the substrate proteins through the specific degradation signals and interact with the Atg8-family proteins to form the autophagosome for degradation [Citation32,Citation33]. The major degradation signal for recognizing cargo receptors in mammals is modifying substrates with ubiquitin [Citation34]. Recent studies have shown that E3 ubiquitin ligase MARCHF8 plays an important role in BST2 degrading MAVS and PEDV N protein [Citation22,Citation32]. To test whether MARCHF8 is involved in regulating LC3 stability mediated by SVA 2AB, co-IP assays were performed with an anti-Flag mAb in 293 T cells transfected with plasmids encoding Flag-2AB or empty vectors. The results showed that SVA 2AB protein interacts with MARCHF8 (). GST affinity-isolation assay indicates that SVA 2AB protein directly binds to MARCHF8 (). We also investigated the colocalization of 2AB-MARCHF8 and found that SVA 2AB protein interacts with MARCHF8 (). We next found that SVA 2AB protein interacts with LC3 ( a nd E), and LC3 protein interacts with MARCHF8 ( a nd G), which suggested that the 2AB, MARCHF8, and LC3 protein combine in pairs to form a large complex for autophagic degradation. Furthermore, we observed that overexpressing MARCHF8 increased the ability of SVA 2AB to degrade the LC3 protein (), and knockdown of MARCHF8 effectively prevented the degradation of LC3 protein induced by 2AB (). The overexpression of 2AB and MARCHF8 significantly increased the polyubiquitination of LC3 protein (), suggesting that MARCHF8 plays a critical role in the 2AB mediated autophagic degradation.
Figure 3. SVA 2AB promotes LC3 protein degradation through MARCHF8 to ubiquitinate LC3. (A) 293 T cells were transfected with plasmids encoding Flag-2AB or empty vectors for 24 h, followed by Co-IP with anti-Flag binding beads and immunoblot analysis with indicated antibodies. ACTB was used as the sample loading control. (B) The GST-MARCHF8 and Flag-2AB proteins were expressed in bacterial strain BL21 (DE3) and purified for the GST affinity-isolation analysis. (C) HeLa cells were transfected with plasmids encoding MYC-MARCHF8 and Flag-2AB, followed by labeling MYC-MARCHF8 and Flag-2AB with a specific primary antibody. The cell nuclei were stained with DAPI. Fluorescent signals were observed using confocal immunofluorescence microscopy. Scale bars: 100 μm. (D) Co-IP assay of cells transfected with plasmids encoding Flag-2AB or empty vector. (E) Immunofluorescence assay of cells transfected with plasmids encoding Flag-2AB and GFP-LC3. (F) Co-IP assay of cells transfected with plasmids encoding Flag-MARCHF8 or empty vector. (G) Immunofluorescence assays of cells transfected with plasmids encoding MYC-MARCHF8 and GFP-LC3. (H) 293 T cells were transfected with plasmids encoding Flag-2AB and MYC-MARCHF8 or empty vectors for 24 h. The protein was harvested for immunoblot analysis. (I) 293 T cells were transfected with plasmids encoding Flag-2AB and MARCHF8 siRNA or negative control (NC) siRNA for 24 h. The protein was harvested for immunoblot analysis. (J) 293 T cells were co-transfected with GFP-LC3 plasmid and Flag-2AB or MYC-MARCHF8 plasmid. Cell lysates were harvested after CQ (10 μM) treatment for 8 h. Proteins extracts were immunoprecipitated using an anti-GFP antibody and analyzed by Western blotting using anti-ubiquitin (Ub) and anti-GFP antibodies.
SVA 2AB promotes the autophagic degradation of MAVS by interaction with MARCHF8
The interferon (IFN) system plays an important role in host defense against viral invasion. Consequently, viruses have developed many strategies to manipulate IFN signaling to establish productive infection [Citation35,Citation36]. To determine whether SVA 2AB protein is involved in regulating the IFN-I signaling pathway, we performed IFNB/IFN-β promoter and IFN-stimulated response element (ISRE)-driven luciferase reporter assay and found that 2AB protein inhibited IFN-I activation in a dose-dependent manner ( a nd B).
Figure 4. SVA 2AB promotes the autophagic degradation of MAVS through combining with MARCHF8. (A and B) 293 T cells were transfected with the IFNB or ISRE luciferase reporter, together with the increasing amounts (wedge) of the expression vector for Flag-2AB. (C) 293 T cells were transfected with the increasing concentrations of plasmids encoding Flag-2AB for 24 h. The cell lysates were analyzed with Western blotting. ACTB was used as the sample loading control. (D) 293 T cells were transfected with plasmids encoding Flag-2AB. Twenty-four hours later, the cells were treated with Baf A1 (0.1 μM), CQ (10 μM), Rapa (6.25 μM), or MG132 (5 μM). The cell lysates were then analyzed by Western blotting. (E) 293 T cells were transfected with plasmids encoding MYC-MAVS and Flag-2AB or empty vectors for 24 h, followed by Co-IP with anti-Flag binding beads and immunoblot analysis with indicated antibodies. (F) HeLa cells were transfected with plasmids encoding MYC-MAVS and Flag-2AB, then labeling MYC-MAVS and Flag-2AB with a specific primary antibody. The cell nuclei were stained with DAPI. Fluorescent signals were observed using confocal immunofluorescence microscopy. Scale bars: 100 μm. (G) The GST-MAVS and Flag-2AB proteins were expressed in bacterial strain BL21 (DE3) and purified for the GST affinity-isolation analysis. (H) 293 T cells were transfected with plasmids encoding Flag-2AB and MYC-MARCHF8 or empty vectors for 24 h. The protein was harvested for immunoblot analysis. (I) 293 T cells were transfected with plasmids encoding Flag-2AB and MARCHF8 siRNA or negative control (NC) siRNA for 24 h. The protein was harvested for immunoblot analysis. Data are means ± SD of triplicate samples. *P < 0.05, **P < 0.01, ***P < 0.001 (two-tailed Student’s t-test).
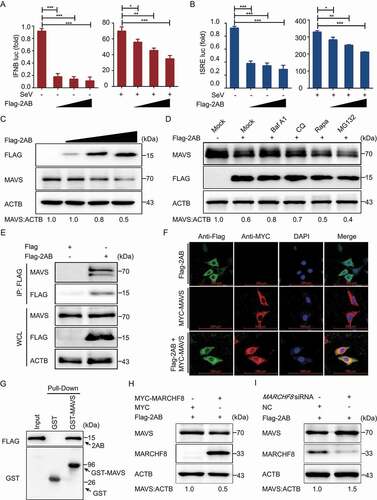
MAVS/Cardiff/IPS-1/VISA (mitochondrial antiviral signaling protein) is mainly involved in inducing IFN-I [Citation37]. A previous study demonstrated that MARCHF8 interacted with MAVS and promoted MAVS degradation to attenuate IFN-I production [Citation32]. Because SVA 2AB could degrade the MARCHF8 protein and MARCHF8 interacted with MAVS, we hypothesized that 2AB could regulate MAVS protein abundance in a MARCHF8-dependent manner. Consistently, we observed that 2AB could reduce MAVS protein abundance in a dose-dependent manner (), and the protein abundance of MAVS increased during autophagy inhibited by the inhibitors (). Furthermore, the co-IP and confocal immunofluorescence assays showed that 2AB protein interacted and colocalized with MAVS in the cytoplasm ( a nd F). Furthermore, the GST affinity-isolation assay also verified the binding between 2AB and MAVS protein. GST-fused to the MAVS (GST-MAVS) bound to the 2AB, but GST protein did not (). We next found that the inhibition of MAVS degradation by 2AB was potentiated in the presence of MARCHF8 (). Conversely, MARCHF8 deficiency significantly abrogated the inhibition of MAVS degradation by 2AB (). Altogether, these results suggested that 2AB negatively regulates MAVS protein in a MARCHF8-dependent manner, thereby regulating IFN-I signaling.
SVA 2AB promotes MARCHF8 degradation to inhibit the IFN-I signaling pathway
Cells were transfected with increasing 2AB plasmids to investigate the underlying mechanism responsible for MARCHF8 degradation. As a result, 2AB downregulated endogenous MARCHF8 in a dose-dependent manner (), and the degradation of MARCHF8 could be blocked by autophagy inhibitors, such as CQ and Baf A1, and promoted by autophagy inducer Rapa (), suggesting that SVA 2AB protein prompts the degradation of MARCHF8 through autophagy. Our findings showed that 2AB protein inhibited IFN-I production and degraded the MARCHF8 protein, indicating that MARCHF8 might potentiate the IFN-I signaling pathway [Citation38,Citation39]. Cells were transfected with MARCHF8 plasmid to test this hypothesis. Interestingly, MARCHF8 increased the IFNB and ISRE luciferase activity than the control (). Conversely, knockdown of MARCHF8 in 293 T cells impaired IFNB promoter activity (). Altogether, these results indicated that MARCHF8 is a positive regulator of the IFN-I signaling pathway.
Figure 5. SVA 2AB promotes MARCHF8 degradation suppressing the activation of IFN-I signaling. (A) 293 T cells were transfected with the increasing concentrations of plasmids encoding Flag-2AB for 24 h. The cell lysates were analyzed with Western blotting. ACTB was used as the sample loading control. (B) 293 T cells were transfected with plasmids encoding Flag-2AB and MYC-MARCHF8. Twenty-four hours later, the cells were treated with Baf A1 (0.1 μM), CQ (10 μM), Rapa (6.25 μM), or MG132 (5 μM). The cell lysates were then analyzed by Western blotting. (C) Lysates of 293 T cells transfected with the IFNB (up) or ISRE (down) luciferase reporter, together with the plasmid encoding MARCHF8 and empty vector, followed by SeV infection, were used for luciferase assay. (D) Lysates of 293 T cells transfected with the IFNB (up) or ISRE (down) luciferase reporter, together with the MARCHF8 siRNA or NC siRNA, followed by SeV infection, were used for luciferase assay. (E and F) PK-15 cells were mock-infected or infected with SVA at an MOI (multiplicity of infection) of 1 and harvested at indicated times. The protein and transcription level of MARCHF8 were analyzed by Western blotting and real-time PCR assays. (G) PK-15 cells were transfected with the vector expressing MARCHF8 or vector control, infected with SVA (MOI = 1). SVA 3D protein was analyzed with Western blotting. (H) The viral RNA levels in the culture supernatants of PK-15 cells treated as described in (G) were determined using real-time PCR. (I and J) PK-15 cells were transfected with MARCHF8 siRNA or negative control siRNA for 24 h. The cells were infected with SVA. Viral protein abundance and viral RNA levels were measured using Western blotting and real-time PCR. (K) Luciferase activity in 293 T cells transfected with IFNB promoter-driven luciferase reporters and together with plasmid encoding Flag-2AB and MYC-MARCHF8 or empty vector. (L) Luciferase activity in 293 T cells transfected with IFNB promoter-driven luciferase reporters and together with plasmid encoding Flag-2AB and MARCHF8 siRNA or negative control siRNA. Data are means ± SD of triplicate samples. *P < 0.05, **P < 0.01, ***P < 0.001 (two-tailed Student’s t-test).
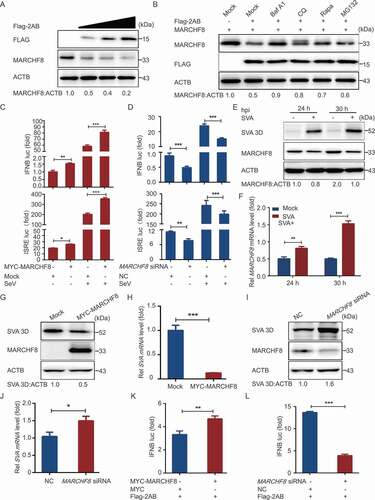
To further investigate whether SVA infection affected MARCHF8 expression, we examined the protein expression level of MARCHF8 in PK-15 cells infected with SVA and harvested at the indicated times. Western blotting showed the decreased abundance of MARCHF8 in SVA-infected PK-15 cells (). Surprisingly, the mRNA level of MARCHF8 was upregulated in the SVA-infected cells (), suggesting that the decrease of MARCHF8 protein level was not due to its change at the transcription level. Next, we evaluated the role of MARCHF8 proteins in SVA infection and found that overexpression of MARCHF8 significantly inhibited SVA replication ( a nd H), and knockdown of endogenous MARCHF8 promoted viral replication ( a nd J). Because SVA 2AB protein inhibited IFN-I activation and prompted the degradation of MARCHF8, and MARCHF8 positively regulated IFN-I signaling, we hypothesized that SVA 2AB might inhibit IFN-I activation by degrading the MARCHF8 protein. We found that overexpression of MARCHF8 markedly induces the IFN-I activation in SVA 2AB transfected cells (), and knockdown of MARCHF8 effectively inhibited IFN-I activation (). Altogether, these results suggested that SVA 2AB degraded the MARCHF8 protein to suppress the activation of IFN-I signaling.
Discussion
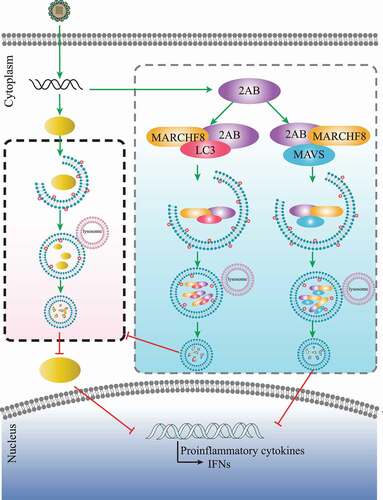
As a newly emerging pathogen, SVA outbreaks in pigs in different regions and countries, resulting in significant economic losses [Citation2]. Thus, with SVA disease appearing in new locations each year, the virus becomes a virus with global prevalence [Citation40]. Autophagy is a cellular physiological process and plays an important role in the antiviral innate immune responses [Citation41,Citation42]. However, little is known regarding the relationship between SVA infection and autophagy. This study demonstrated that SVA-induced autophagy inhibited viral replication by degrading the SVA 3 C protein in the early stage of viral infection, and SVA 2AB protein inhibited autophagy through MARCHF8 to ubiquitinate LC3 in the late stage of viral infection. During SVA infection, SVA 2AB protein also promotes MARCHF8 and MAVS degradation to inhibit IFN-I signaling ().
Figure 6. A proposed working model to illustrate autophagy inhibited SVA replication by degrading virus 3C protein, and 2AB protein degrading LC3, MARCHF8 and MAVS antagonize selective autophagy and type I interferon production in SVA infection.
Autophagy is a conserved degradation pathway that damages organelles and misfolded or long-lived proteins to maintain cellular homeostasis. During selective autophagy, the damaged proteins would be modified with ubiquitin and then recognized by cargo receptors. The complexes of cargo receptors and specific substrates interact directly with the Atg8-family proteins and are sequestered within double-membrane vesicles called “autophagosomes”, which fuse with lysosomes. The inner autophagosomal membrane, cargo receptors, and specific substrates are degraded in the lysosome’s proteolytic environment [Citation43]. Besides the physiological functions in cellular homeostasis, autophagy has been shown to play an important role in immune response and viral replication [Citation5,Citation44]. For instance, autophagy induced by transmissible gastroenteritis virus/TGEV infection suppresses viral replication [Citation45]. In addition, autophagy was confirmed to inhibit Japanese encephalitis virus/JEV replication [Citation41]. Previous studies reported that autophagy can be induced by SVA infection and promotes viral replication in pig cells [Citation11,Citation28].
Interestingly, we found that SVA-induced autophagy inhibited viral replication in the early stage of viral infection, and the induction of autophagy inhibited SVA replication in PK-15 cells. SVA 3 C protein has been reported to regulate the host antiviral immunity through different strategies negatively. 3 C protein negatively regulates the type I interferon pathway by degrading IRF3, IRF7, and DDX58/RIG-I and targets MAVS, TRIM69/TRIF, and TANK for cleavage through its protease activity [Citation14–16]. The 3 C protein also could disrupt the EIF4G1-G3BP1 interaction to inhibit stress granule formation [Citation46]. During virus infection, the induced autophagy could degrade the viruses and viral-derived antigens to inhibit virus replication. It has been reported that the host factors BST2 and IRAV could induce selective autophagy to degrade the PEDV N protein and reduce PEDV replication [Citation22,Citation42]. We found that the SVA 3 C protein was degraded by Rapa-induced autophagy, suggesting that SVA-induced autophagy might inhibit viral replication via degradation of SVA 3 C.
As autophagy contributes to host immunity and inhibits viral replication, viruses have evolved various strategies to counteract or hijack the autophagy process to facilitate viral replication. Previous research shows that autophagy negatively regulates Japanese encephalitis virus replication during the early stages of infection and becomes dysfunctional as the infection progresses [Citation41]. Here, we demonstrated that autophagy inhibited in the late stage of SVA infection (after 12 h). Similar to other picornaviruses, the polyprotein precursor of SVA can be stepwisely cleaved into different polypeptides [Citation3]. The nonstructural protein 2A only contains nine amino acids is much smaller than the 2A protein of other picornaviruses, which may lose some protein function compared with other picornaviruses 2A proteins. This study has shown that SVA 2AB protein plays an important role in inhibiting autophagy. However, overexpressed 2A or 2B protein alone in cells could not inhibit the autophagy progress (data not shown). Therefore, although the polyprotein precursor of picornaviruses can be cleaved into different proteins, in which 2A and 2B are two independent proteins that play different functions, respectively, the precursor protein 2AB of SVA might play a role in inhibiting autophagy before cleavage or 2AB protein might not be cleaved into 2A and 2B, but instead functions as a single protein during SVA replication. In autophagy, some substrate proteins must be ubiquitinated to serve as a major signal responsible for recognizing cargo receptors. Previous studies revealed that E3 ubiquitin ligase MARCHF8 catalyzed the ubiquitination of the PEDV N protein during IRAV and BST2 degradation of N protein [Citation22,Citation42]. Here, we found that 2AB, MARCHF8, and LC3 protein combine in pairs, and changing the protein abundance of MARCHF8 affected the stability of the LC3 protein. Our findings prove that MARCHF8 plays a critical role in the SVA 2AB protein-mediated autophagic degradation. 2AB combined with MARCHF8 and LC3 and facilitated the MARCHF8-mediated polyubiquitination of the LC3 protein. So, we speculated that the ubiquitinated LC3 protein was transferred into the autophagosomal membrane and degraded in the lysosome, and this process may not require the cargo receptors.
Antiviral responses are mediated primarily by the IFN-I, triggering the production of hundreds of interferon-stimulating genes (ISGs) to establish an antiviral state. Consequently, viruses have developed lots of strategies to manipulate IFN signaling to establish a productive infection. During SVA infection, the 3 Cpro plays an important role in antagonizing the IFN-I signaling pathway basis on its protease properties [Citation15,Citation16,Citation28]. Here, we found that SVA 2AB protein is a negative regulator of the IFN-I signaling pathway. Previous studies have shown that selective autophagy regulates the stability of transcription factor IRF3 to balance IFN-I production and immune suppression [Citation44]. Tetherin recruits MARCHF8 catalyzing ubiquitin chains on MAVS for selective autophagy degradation to antagonize the IFN response [Citation32]. Our finding proved that SVA 2AB protein negatively regulates the MAVS protein in a MARCHF8-dependent manner. Overexpressed MARCHF8 increased the IFNB and ISRE luciferase activity, and the knockdown of MARCHF8 impaired the IFN-I signaling pathway, indicating that MARCHF8 is a positive regulator of the IFN-I signaling pathway. During SVA infection, SVA 2AB, MARCHF8, and LC3 protein combine in pairs packaging into an autophagosome for degradation and interrupting MARCHF8 expression effectively inhibited the IFN-I activation in SVA 2AB transfected-cells. Therefore, SVA 2AB degraded MARCHF8 protein to suppress the activation of the IFN-I signaling pathway.
In summary, we found that SVA-induced autophagy inhibited viral replication by degrading the SVA 3 C protein in the early stage of viral infection. SVA 2AB protein inhibited autophagy through MARCHF8 to ubiquitinate LC3 in the late stage of viral infection, promoting SVA replication. We also illustrated a new function of MARCHF8 as a positive regulator of IFN-I signaling, and SVA 2AB protein promoted MARCHF8 and MAVS degradation to inhibit IFN-I signaling during SVA infection. Our findings provide insight into the role of autophagy and anti-innate immune strategies to manipulate IFN signaling during SVA replication.
Materials and methods
Antibodies, reagents, and plasmids
Anti-LC3A/B antibody (12,741), anti-HA-tag antibody (3724), anti-GFP-tag antibody (55,494), and anti-MYC antibody (2276) were purchased from Cell Signaling Technology. Anti-Flag M2 antibody produced in mouse (F1804) was obtained from Sigma-Aldrich. Anti-MAVS (SC-166583) and anti-ubiquitin antibody (SC-8017) were purchased from Santa Cruz Biotechnology. Anti-MARCHF8/MARCH8 antibody (14,119-1-AP), anti-GST-tag antibody (10,000-0-AP), anti-ACTB/β-actin antibody (60,008–1), horseradish peroxidase (HRP)-conjugated anti-mouse IgG antibody (SA00001-1), and horseradish peroxidase (HRP)-conjugated anti-rabbit IgG antibody (SA00001-2) were purchased from the Proteintech Group. Polyclonal antibody against SVA (GD-2018) 3D protein was prepared in our laboratory. Bafilomycin A1 (Baf A1; 54,645) was purchased from Cell Signaling Technology. MG132 (M7449) and chloroquine phosphate (CQ; PHR1258) were purchased from Sigma-Aldrich. Rapamycin (HY-10219) was purchased from MedChemExpress (Shanghai, China). The Dual-Glo Luciferase Assay System (DL101) and ClonExpress II One Step Cloning Kit (C112-02) were purchased from Vazyme Biotech Co.,ltd. MARCHF8 siRNA were designed and purchased from GenePharma (Shanghai, China) as described previously [Citation22]. The plasmid of GFP-LC3 was kindly provided by HongChao Gou (Institute of Animal Health, Guangdong Academy of Agricultural Sciences, Guangzhou, China).
Cell cultures, transfection, and viral infection
Porcine kidney cells (PK-15 cells; ATCC, CCL-33) and human embryonic kidney cells (293 T cells; ATCC, CRL-11268) were cultured in DMEM (Sigma-Aldrich, D6429) supplemented with 10% heat-inactivated FBS (Gibco, 10,099,141) at 37°C under 5% CO2 in a humidified incubator. Cells transfection was performed using Lipofectamine 3000 Reagent (Invitrogen, L3000015) according to procedures recommended by the manufacturer. According to the manufacturer’s instructions, siRNA duplexes were transfected using Lipofectamine RNAiMAX (Invitrogen, 13,778,150). The SVA strain (GD-2018) was isolated and stored in our laboratory. SVA was propagated and titrated using PK-15 cells. The viral titers were calculated using Karber’s method and expressed as 50% tissue culture infective dose (TCID50) [Citation47].
Western blotting analysis
Cells were incubated on ice with RIPA Lysis and Extraction Buffer (Thermo Fisher Scientific, 89,901) containing Protease Inhibitor Cocktail (Bimake, B14001) and Phosphatase Inhibitor Cocktail (Bimake, B15001) for 5 min. The lysates were denatured in 5 × SDS-PAGE Sample loading buffer (Beyotime Biotechnology, P0015) for 10 min and separated with SDS-PAGE, then the proteins were transferred onto nitrocellulose Western blotting membranes (GE Healthcare, 10,600,001). The membranes were blocked for 1 h at room temperature (RT) with 5% nonfat dry milk (BD, 232,100) and incubated with the primary antibody at RT for 1 h. After washing with PBS (Gibco, C20012500BT) containing 0.1% Tween 20 (Sigma-Aldrich, P1379), the membranes were incubated with the corresponding HRP-conjugated secondary antibody at RT for 1 h and detected with enhanced chemiluminescence (ECL; Share-bio, SB-WB012).
Co-immunoprecipitation assay
Cells were transfected with the indicated plasmids for 24 h and incubated on ice NP40 Cell Lysis Buffer (Life Technologies, FNN0021) containing Protease Inhibitor Cocktail and Phosphatase Inhibitor Cocktail for 30 min. The lysates were centrifuged and incubated with affinity antibodies bound to Dynabeads Protein G (Life Technologies, 10004D) for 30 min at RT. Subsequently, the Dynabeads were washed with PBS containing 0.02% Tween 20 and boiled in sample buffer. The proteins were analyzed with immunoblotting using the indicated antibodies.
RNA extraction and quantitative real-time PCR
Total RNA was extracted from cells using the QIAamp Viral RNA Mini Kit (Qiagen, 52,906) according to the manufacturer’s instructions, and One microgram of the total RNA was reverse-transcribed using PrimeScript™ RT reagent Kit (TaKaRa Bio, RRO47A). The real-time RT-PCR was performed using the SYBR Premix Ex Taq™ (Vazyme Biotech Co., ltd, q711-03). The primer sequences used for the qRT-PCR analysis are listed in Table S1. All data were normalized to ACTB or GAPDH expression.
GST affinity-isolation assay
The full-length MARCHF8 gene, MAVS gene, and Flag-2AB gene were cloned into pCold GST plasmid (Clontech Laboratories, Inc, 3372) or pCold TF plasmid (Clontech Laboratories, Inc, 3365). Recombinant proteins were expressed in BL21 competent cells (Vazyme Biotech Co., ltd, C504-03). According to the manufacturer’s instructions, the interaction of MARCHF8 and Flag-2AB protein or MAVS and Flag-2AB protein was analyzed by the GST Protein Interaction Pull-Down Kit (Thermo Fisher Scientific, 21,516). The bound proteins were eluted by reduced glutathione and analyzed with Western blotting using the indicated antibodies.
Confocal immunofluorescence assay
Cells were cultured on coverslips and transfected with the specific expression plasmids for 24 h. Cells were fixed with 4% paraformaldehyde (Sigma-Aldrich, P6148) for 15 min and permeabilized with 0.1% Triton X-100 (Sigma-Aldrich, T9284) at RT for 10 min. The cells were blocked with 5% bovine serum albumin (Cell Signaling Technology, 9998) for 1 h and then incubated with primary antibody for 1 h at 37°C. After three washes with PBS, the cells were incubated with fluorescently labeled secondary antibody for 1 h at 37°C in the dark. Nuclei were stained with 4′,6-diamidino-2-phenylindole (Beyotime Biotechnology, C1002) for 5 min. Fluorescence images were acquired with confocal immunofluorescence microscopy (Carl Zeiss, Oberkochen, Germany).
Cell viability assay
Cell viability was determined by the MTT assay as described previously [Citation5]. Cells were seeded in 96-well culture plates and cultured for 24 h at 37°C. Then, the fresh medium containing the corresponding concentrations of pharmacological drugs was replaced, and the cells were continuously incubated for 24 h. Next, MTT (Abcam, ab211091) was added to the culture medium and inoculated for 4 h. Then, the formazan solution was added to each well, and a microplate spectrophotometer measured the optical density at 490 nm.
Statistical analysis
All experiments were repeated at least three times independently. Data were analyzed with the two-tailed Student’s t-test using GraphPad Prism 5 software (GraphPad Software, USA) and presented as means ± standard deviations (SD). P-value < 0.05 were considered statistically significant.
Supplemental Material
Download Zip (15.6 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplemental Material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Hales LM, Knowles NJ, Reddy PS, et al. Complete genome sequence analysis of Seneca Valley virus-001, a novel oncolytic picornavirus. J Gen Virol. 2008;89(5):1265–1275.
- Zhang X, Zhu Z, Yang F, et al. Review of Seneca Valley virus: a call for increased surveillance and research. Front Microbiol. 2018;9:940.
- Liu F, Wang Q, Huang Y, et al. A 5-year review of Senecavirus A in China since its emergence in 2015. Front Vet Sci. 2020;7:567792.
- Oliveira TES, Michelazzo MMZ, Fernandes T, et al. Histopathological, immunohistochemical, and ultrastructural evidence of spontaneous Senecavirus A-induced lesions at the choroid plexus of newborn piglets. Sci Rep. 2017;7(1):16555.
- Sun P, Zhang S, Qin X, et al. Foot-and-mouth disease virus capsid protein VP2 activates the cellular EIF2S1-ATF4 pathway and induces autophagy via HSPB1. Autophagy. 2018;14(2):336–346.
- Zhang J, Zhang H, Sun W, et al. Genetic evolution and epidemiological analysis of Seneca Valley virus (SVV) in China. Virus Res. 2021;291:198177.
- Pasma T, Davidson S, Shaw SL, et al. Idiopathic vesicular disease in swine in Manitoba. Can Vet J. 2008;49(1):84–85.
- Guo B, Pineyro PE, Rademacher CJ, et al. Novel Senecavirus A in swine with vesicular disease, United States, july 2015. Emerg Infect Dis. 2016;22(7):1325–1327.
- Saeng-Chuto K, Rodtian P, Temeeyasen G, et al. The first detection of Senecavirus A in pigs in Thailand, 2016. Transbound Emerg Dis. 2018;65(1):285–288.
- Vannucci FA, Linhares DC, Barcellos DE, et al. Identification and complete genome of Seneca Valley virus in vesicular fluid and sera of pigs affected with idiopathic vesicular disease, Brazil. Transbound Emerg Dis. 2015;62(6):589–593.
- Wu Q, Zhao X, Bai Y, et al. The first identification and complete genome of Senecavirus A affecting pig with idiopathic vesicular disease in China. Transbound Emerg Dis. 2017;64(5):1633–1640.
- Willcocks MM, Locker N, Gomwalk Z, et al. Structural features of the Seneca Valley virus internal ribosome entry site (IRES) element: a picornavirus with a pestivirus-like IRES. J Virol. 2011;85(9):4452–4461.
- Yang X, Hu Z, Fan S, et al. Picornavirus 2A protease regulates stress granule formation to facilitate viral translation. PLoS Pathog. 2018;14(2):e1006901.
- Qian S, Fan W, Liu T, et al. Seneca Valley virus suppresses host type i interferon production by targeting adaptor proteins MAVS, TRIF, and TANK for cleavage. J Virol. 2017;91.
- Xue Q, Liu H, Zhu Z, et al. Seneca Valley Virus 3Cpro abrogates the IRF3- and IRF7-mediated innate immune response by degrading IRF3 and IRF7. Virology. 2018;518:1–7.
- Wen W, Yin M, Zhang H, et al. Seneca Valley virus 2C and 3C inhibit type I interferon production by inducing the degradation of RIG-I. Virology. 2019;535:122–129.
- Xue Q, Liu H, Zhu Z, et al. Seneca Valley virus 3Cpro cleaves PABPC1 to promote viral replication. Pathogens. 2020;9(6):443.
- Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290(5497):1717–1721.
- Boya P, Reggiori F, Codogno P, et al. Emerging regulation and functions of autophagy. Nat Cell Biol. 2013;15(7):713–720.
- Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306(5698):990–995.
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42.
- Kong N, Shan T, Wang H, et al. BST2 suppresses porcine epidemic diarrhea virus replication by targeting and degrading virus nucleocapsid protein with selective autophagy. Autophagy. 2020;16(10):1737–1752.
- Orvedahl A, Alexander D, Talloczy Z, et al. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1(1):23–35.
- Lussignol M, Queval C, Bernet-Camard M-F, et al. The herpes simplex virus 1 Us11 protein inhibits autophagy through its interaction with the protein kinase PKR. J Virol. 2013;87(2):859–871.
- Liu G, Zhong M, Guo C, et al. Autophagy is involved in regulating influenza A virus RNA and protein synthesis associated with both modulation of Hsp90 induction and mTOR/p70S6K signaling pathway. Int J Biochem Cell Biol. 2016;72:100–108.
- Lee Y-R, Lei H-Y, Liu M-T, et al. Autophagic machinery activated by dengue virus enhances virus replication. Virology. 2008;374(2):240–248.
- Pei J, Zhao M, Ye Z, et al. Autophagy enhances the replication of classical swine fever virus in vitro. Autophagy. 2014;10(1):93–110.
- Wen W, Li X, Yin M, et al. Selective autophagy receptor SQSTM1/ p62 inhibits Seneca Valley virus replication by targeting viral VP1 and VP3. Autophagy. 2021:1–13.
- Hou L, Dong J, Zhu S, et al. Seneca valley virus activates autophagy through the PERK and ATF6 UPR pathways. Virology. 2019;537:254–263.
- Wu Y, Yang X, Yao Z, et al. C19orf66 interrupts Zika virus replication by inducing lysosomal degradation of viral NS3. PLoS Negl Trop Dis. 2020;14(3):e0008083.
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–741.
- Jin S, Tian S, Luo M, et al. Tetherin suppresses type I interferon signaling by targeting MAVS for NDP52-mediated selective autophagic degradation in human cells. Mol Cell. 2017;68(2):308–22 e4.
- Kraft C, Peter M, Hofmann K, et al. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol. 2010;12(9):836–841.
- Deretic V, Saitoh T, Akira S, et al. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13(10):722–737.
- Ding Z, Fang L, Jing H, et al. Porcine epidemic diarrhea virus nucleocapsid protein antagonizes beta interferon production by sequestering the interaction between IRF3 and TBK1. J Virol. 2014;88:8936–8945.
- Guo L, Luo X, Li R, et al. Porcine epidemic diarrhea virus infection inhibits interferon signaling by targeted degradation of STAT1. J Virol. 2016;90:8281–8292.
- Seth RB, Sun L, Ea CK, et al. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682.
- Zheng C, Tang YD. When MARCH family proteins meet viral infections. Virol J. 2021;18:49.
- Zheng C. The emerging roles of the MARCH ligases in antiviral innate immunity. Int J Biol Macromol. 2021;171:423–427.
- Houston E, Temeeyasen G, Pineyro PE, et al. Comprehensive review on immunopathogenesis, diagnostic and epidemiology of Senecavirus A. Virus Res. 2020;286:198038.
- Sharma M, Bhattacharyya S, Nain M, et al. Japanese encephalitis virus replication is negatively regulated by autophagy and occurs on LC3-I- and EDEM1-containing membranes. Autophagy. 2014;10:1637–1651.
- Wang H, Kong N, Jiao Y, et al. EGR1 suppresses porcine epidemic diarrhea virus replication by regulating IRAV to degrade viral nucleocapsid protein. J Virol. 2021;JVI0064521.
- Paul P, Munz C. Autophagy and mammalian viruses: roles in immune response, viral replication, and beyond. Adv Virus Res. 2016;95:149–195.
- Wu Y, Jin S, Liu Q, et al. Selective autophagy controls the stability of transcription factor IRF3 to balance type I interferon production and immune suppression. Autophagy. 2021;17:1379–1392.
- Guo L, Yu H, Gu W, et al. Autophagy negatively regulates transmissible gastroenteritis virus replication. Sci Rep. 2016;6:23864.
- Wen W, Zhao Q, Yin M, et al. Seneca Valley virus 3C protease inhibits stress granule formation by disrupting eIF4GI-G3BP1 interaction. Front Immunol. 2020;11:577838.
- Kong N, Wu Y, Meng Q, et al. Suppression of virulent porcine epidemic diarrhea virus proliferation by the PI3K/Akt/GSK-3alpha/beta pathway. PloS One. 2016;11:e0161508.