
ABSTRACT
High levels of reactive oxygen species (ROS) result in oxidative stress, which damages cells and leads to the development of many diseases. Macroautophagy/autophagy plays an important role in protecting cells from diverse stress stimuli including oxidative stress. However, the molecular mechanisms of autophagy activation in response to oxidative stress remain largely unclear. In this study, we showed that TRAF6 mediates oxidative stress-induced ATG9A ubiquitination at two C-terminal lysine residues (K581 and K838). ATG9A ubiquitination promotes its association with BECN1, BECN1-PIK3C3/VPS34-UVRAG complex assembly and PIK3C3/VPS34 activation, thereby activating autophagy and endocytic trafficking. We also identified TNFAIP3/A20 as a negative regulator of oxidative-induced autophagy by counteracting TRAF6-mediated ATG9A ubiquitination. Moreover, ATG9A depletion attenuates LPS-induced autophagy and causes aberrant TLR4 signaling and inflammatory responses. Our findings revealed a critical role of ATG9A ubiquitination in oxidative stress-induced autophagy, endocytic trafficking and innate immunity.
Reactive oxygen species (ROS) are highly reactive oxygen-containing molecules that play important roles in the regulation of cell signaling and homeostasis. Cellular ROS are mainly derived from NADPH oxidases (NOXs) and mitochondria. However, excessive levels of ROS cause oxidative stress and cellular damage, which lead to a variety of human diseases. Diverse cellular processes contribute to protect cell survival in response to oxidative stress. Macroautophagy (hereafter autophagy) is a cellular protective mechanism that degrades and recycles damaged cell components in response to stress conditions, including oxidative stress. The autophagy pathway proceeds through a series of highly regulated steps, including initiation, membrane nucleation, expansion and closure of the nascent autophagosomal membrane, and finally fusion with a lysosome for cargo degradation. Nevertheless, the molecular mechanisms underlying the activation of oxidative stress-mediated autophagy remain elusive.
ATG9 is a multi-spanning transmembrane protein that cycles between different cellular compartments, including the trans-Golgi network (TGN), endosomes and autophagic membranes. Recent studies have indicated a critical role of ATG9 in autophagosome formation by mediating lipid scrambling during the expansion of the phagophore membrane. In this study, we found that mammalian ATG9A is markedly ubiquitinated in response to oxidative stress but not to nutrient starvation or ER stress [Citation1]. We further demonstrated that the E3 ubiquitin ligase TRAF6 mediates nonproteolytic ubiquitination of ATG9A upon oxidative stress. TRAF6 is essential for H2O2-induced autophagy activation and ATG9A localization on nascent autophagosomes. TRAF6 E3 ubiquitin ligase mainly promotes K63-linked polyubiquitination. Interestingly, we found overexpression of TRAF6 or H2O2 treatment induces K48- and K63-linked polyubiquitination of ATG9A (). However, the topology of the K48- and K63-linked polyubiquitination on ATG9A and whether E3 ubiquitin ligases other than TRAF6 are involved in mediating ATG9A ubiquitination remain to be elucidated.
Figure 1. Scheme depicting the mechanisms of ATG9A ubiquitination in oxidative stress induced-autophagy. TNFAIP3/A20 interacts with ATG9A under normal conditions. Upon oxidative stress, TRAF6 mediates ATG9A ubiquitination, thereby enhancing ATG9A-BECN1 interaction, BECN1-PIK3C3/VPS34-UVRAG complex assembly, PIK3C3/VPS34 activation, autophagosome formation, and endocytic trafficking. Ablation of TRAF6 or ATG9A, or expression of TNFAIP3/A20 or ATG9A ubiquitination mutant impairs BECN1-PIK3C3/VPS34-UVRAG complex assembly and PIK3C3/VPS34 activity.
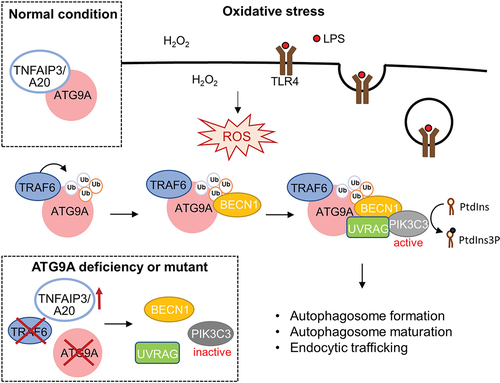
Ubiquitination is one of the most important protein posttranslational modifications that has been implicated in the regulation of substrate activity, cellular localization, protein stability, and interactions with other proteins. Notably, we found that TRAF6-mediated ATG9A ubiquitination enhances its interaction with another key autophagy initiator, BECN1 (beclin 1) during oxidative stress. BECN1 acts as a scaffolding protein that recruits various interacting partners to form multiple PIK3C3/VPS34 complexes with distinct functions. The class III phosphatidylinositol 3-kinase (PtdIns3K) PIK3C3/VPS34 phosphorylates the membrane lipid phosphatidylinositol (PtdIns) to PtdIns3P and is critical for both autophagosome formation and endocytic trafficking. BECN1-PIK3C3/VPS34 interacts with ATG14 and UVRAG to form PtdIns3K complex I and complex II, respectively, to regulate autophagy at different stages. Interestingly, we found H2O2 treatment dramatically enhanced the association between ATG9A and UVRAG and BECN1-PIK3C3/VPS34-UVRAG complex formation. Significantly, ATG9A ablation attenuates BECN1-PIK3C3/VPS34-UVRAG complex assembly and PIK3C3/VPS34 activity. The BECN1-PIK3C3/VPS34-UVRAG complex has been reported to be involved in autophagosome maturation and endocytic trafficking. Consistent with our findings, ATG9A knockdown also impairs autophagosome maturation and endocytic trafficking.
Using mass spectrometry (MS)-based analysis, we identified two highly conserved C- terminal lysine residues (K581 and K838) of ATG9A as the TRAF6-mediated ubiquitination sites. The ATG9AK581R,K838R double mutant attenuates oxidative stress-induced interaction with BECN1, PIK3C3/VPS34 activity, autophagy flux and endocytic trafficking, indicating that the ubiquitination of ATG9A at K581 and K838 is critical for its function to regulate autophagy in response to oxidative stress. However, the molecular interaction between ATG9A ubiquitination and the BECN1-PIK3C3/VPS34-UVRAG complex in oxidative stress-induced autophagy and endocytic trafficking remains to be further clarified.
Like many types of posttranslational modifications, ubiquitination is a reversible process. Ubiquitin modification can be catalyzed by E3 ubiquitin ligases and reversed by the removal of ubiquitin chains via deubiquitinases (DUBs). The DUB proteins play important roles in the control of the ubiquitin signaling network, thereby maintaining the homeostasis of various cellular processes. In this study, we found that ATG9A interacts with the OTU family deubiquitinase TNFAIP3/A20 under normal conditions, whereas their interaction dramatically decreases upon oxidative stress stimulation. Moreover, TNFAIP3/A20 attenuates TRAF6- and H2O2-induced ATG9A ubiquitination and the interaction between ATG9A and BECN1. These results suggest that TNFAIP3/A20 coordinates with TRAF6 to regulate ATG9A ubiquitination and autophagy under oxidative stress conditions. Our study demonstrates how the dynamic ubiquitination of ATG9A plays a critical role in the regulation of autophagy in response to oxidative stress.
Various biological processes generate ROS and cause intracellular oxidative stress. ROS production is essential for immune response activation and pathogen elimination; however, elevated levels of ROS also cause cell injury and cell death. Lipopolysaccharide (LPS) is the major surface membrane component of Gram-negative bacteria. LPS triggers immune responses and induces oxidative stress through TLR4 (toll like receptor 4)-mediated signaling which also induces autophagy. Here we found that LPS-induced ROS production enhances TRAF6-mediated ATG9A ubiquitination, ATG9A-BECN1 interaction and BECN1-PIK3C3/VPS34-UVRAG complex formation. Moreover, ATG9A depletion markedly reduces BECN1-PIK3C3/VPS34-UVRAG complex formation and autophagy levels upon LPS stimulation, thus suggesting that ATG9A also plays a crucial role in LPS-induced autophagy.
Increasing evidence has indicated the involvement of autophagy in the regulation of LPS-induced TLR4-mediated innate immunity. Upon LPS binding to plasma membrane TLR4, the inflammatory responses are stimulated through the activation of the NFKB and MAPK (mitogen-activated protein kinase) signaling pathways. In addition, the activated TLR4 can be endocytosed and transported to the endosomal compartments where TBK1 (TANK binding kinase 1) signaling is activated and mediates the phosphorylation and the activation of transcription factor IRF3 (interferon regulatory factor 3) and expression of type I interferon (IFN). We found that LPS-induced IRF3 phosphorylation is markedly reduced in ATG9A-depleted and 3-MA-treated macrophages, suggesting that ATG9A and PIK3C3/VPS34 activity are essential in the regulation of endosomal TLR4 signaling. Moreover, we found that ATG9A depletion and PIK3C3/VPS34 inhibition dramatically decrease the size of LPS-TLR4+ endosomes but not TLR4 internalization. The increased size of LPS-TLR4-containing early endosomes is likely resulting from endosome-endosome fusion, and UVRAG promotes endosome-endosome fusion. These results together suggest that ATG9A-mediated BECN1-PIK3C3/VPS34-UVRAG complex formation and PIK3C3/VPS34 activation are crucial for LPS-induced TLR4 endosomal signaling. We further showed that ATG9A negatively regulates LPS-induced inflammasome activation. Our study indicates that ATG9A plays a crucial role in LPS-induced inflammatory responses.
Disclosure statement
No potential conflict of interest was reported by the authors
Additional information
Funding
Reference
- Wang Y-T, Liu T-Y, Shen C-H, et al. K48/K63-linked polyubiquitination of ATG9A by TRAF6 E3 ligase regulates oxidative stress-induced autophagy. Cell Rep. 2022;38(8):110354.