
ABSTRACT
The assembly of the NLRP3 inflammasome can be initiated by a wide range of stimuli including exogenous infection as well as endogenous damage. Therefore, the tight regulation of the NLRP3 inflammasome is crucial for the host to resist microbial invasion and maintain homeostasis. Our recent work has identified a negative regulator of NLRP3-mediated inflammation, namely CCDC50 (coiled-coil domain containing protein 50). CCDC50 can be induced by NLRP3 agonists and then functions as a macroautophagy/autophagy cargo receptor to recognize K63-polyubiquitinated NLRP3 and deliver it to MAP1LC3/LC3-conjugated phagophores for degradation. CCDC50 inhibits the polymerization of NLRP3 and the recruitment of PYCARD/ASC, consequently suppressing the assembly of inflammasomes. ccdc50-knockout mice are more susceptible to dextran-sulfate (DSS)-induced colitis and exhibit more severe gut inflammation with elevated NLRP3 inflammasome activity, suggesting a protective role of CCDC50 in the pathology and progression of inflammatory bowel disease (IBD). Our finding reveals a function of autophagy-related proteins in the regulation of NLRP3-mediated inflammation, thus demonstrating the intricate crosstalk between autophagy and inflammation.
The NLRP3 inflammasome is an intracellular multiprotein complex, consisting of one sensor (NLRP3), one adaptor (PYCARD/ASC) and one effector (CASP1 [caspase 1]). Upon activation by various stimuli, NLRP3 undergoes polyubiquitination and oligomerization and then recruits PYCARD. The latter also oligomerizes and forms the structures of a filament and then a speck. PYCARD recruits the effector CASP1 and leads to its self-cleavage and activation. Activated CASP1 cleaves pro-IL1B/IL-1β, pro-IL18 and GSDMD (gasdermin D), initiating inflammatory responses and even pro-inflammatory cell death. Timely activation of the NLRP3 inflammasome can protect host cells from microbial infection, whereas continuous hyperactivation of NLRP3 inflammasome may result in overt cell death and inflammatory diseases including IBD. Autophagy is an evolutionarily conserved process that sequesters intracellular materials including misfolded proteins, aging or damaged organelles and invading pathogens in a phagophore, which then matures into an autophagosome. Autophagic regulation of inflammasome activity is critical in immune homeostasis and autophagy defects have been implicated in excessive activation of the NLRP3 inflammasome and the pathogenesis of autoinflammatory diseases. In our recent study, we reveal an unexpected and previously unknown function of CCDC50, a novel autophagy cargo receptor identified by our group, in NLRP3 inflammasome regulation and autoinflammatory diseases [Citation1].
Our previous study used a genome-wide CRISPR-Cas9-based library to screen the cellular negative regulatory factors involved in type I interferon (IFN) responses and identified CCDC50 as a negative regulator of IFN signaling. Further validation experiments reveal that CCDC50 is a novel autophagy receptor, recognizing K63-polyubiquitin-conjugated DDX58/RIG-I-like receptors (RLRs) and STING1 and targeting them for autophagic degradation. CCDC50 is ubiquitously expressed in several organs but highly enriched in immune tissues and cells, especially in dendritic cells. CCDC50 deficiency causes the enrichment of autoinflammatory disease-related genes and upregulation of autoimmune-related signaling pathways in our RNA-seq data. We find that CCDC50 is downregulated in systemic lupus erythematosus (SLE) and involved in negative regulation in SLE disease progression because of its inhibition of the CGAS-STING1 signaling pathway.
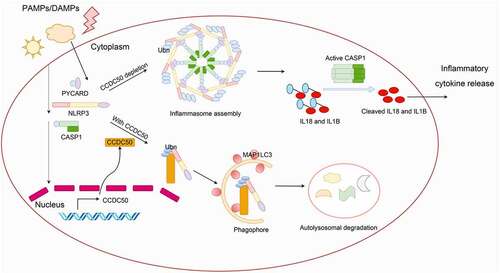
We also find that the activation and assembly of the NLRP3 inflammasome is upregulated in CCDC50-deficient cells. Moreover, the downstream proinflammatory cytokines such as IL1 are also enriched in CCDC50-deleted cells, indicating that CCDC50 might be involved in NLRP3 inflammasome activity. We then isolated bone marrow-derived macrophages (BMDMs) and dendritic cells (BMDCs) and depleted Ccdc50 expression, finding that Ccdc50 deficiency leads to enhanced proinflammatory cytokine responses triggered by a variety of extracellular and intracellular NLRP3 stimuli. Notably, the agonists for NLRP3 inflammasome can induce a slight but significant elevation of CCDC50 mRNA level, suggesting that CCDC50 can be induced by danger/damage-associated molecular patterns to a certain extent. ccdc50-knockout cells and mice were constructed to verify its physiological roles. The data show that CCDC50 deficiency causes enhanced cleavage of pro-CASP1 and increased secretion of IL1B both in human and mouse primary immune cells, demonstrating that CCDC50 inhibits the activation of the NLRP3 inflammasome and IL1B production. We then explored the molecular mechanism behind which CCDC50 negatively regulates NLRP3-depedent inflammation. Exogenous interaction and endogenous association demonstrate that CCDC50 can interact with and further affect the stability of NLRP3. Cycloheximide chase experiments and treatment with inhibitors show that NLRP3 is taken as cargo via CCDC50 to autolysosomes for degradation. K63-linked polyubiquitination of NLRP3 is indispensable for the recognition of CCDC50. During this process, the canonical autophagy machinery is required while SQSTM1/p62 is not necessary. Furthermore, CCDC50 also inhibits the oligomerization of NLRP3, the recruitment of PYCARD and then the formation of PYCARD specks, leading to the inhibition of NLRP3 inflammasome assembly ().
Figure 1. The schematic diagram of a working model of CCDC50 in the regulation of NLRP3 inflammasome activation. Upon PAMPs or DAMPs stimulation, the NLRP3 inflammasome assembly is initiated to assist host cells fight against microbial infection or monitor cellular damage. In the meantime, the autophagy cargo receptor CCDC50 is induced and then CCDC50 targets K63-polyubiqutinated NLRP3 for autophagic degradation, consequently disrupting NLRP3 oligomerization and further inhibiting NLRP3 inflammasome assembly. Ubn, polyubiquitin.
We further employed a DSS-induced mouse colitis model to demonstrate the pathological significance of the regulation of the NLRP3 inflammasome by CCDC50-mediated autophagy. We find that DSS treatment can induce the expression of Ccdc50 in wild-type mice, whereas ccdc50-knockout mice show more severe gut inflammation with increased body weight loss, shortened colon length, more colonic tissue lesions and more inflammatory cell infiltration compared with wild-type littermates. Moreover, the colons isolated from ccdc50-knockout mice show much more oligomerization of NLRP3 and higher production of IL1B than those of wild-type mice. All these findings suggest that upregulated CCDC50 functions as a guardian to suppresses the over-activation of the NLRP3 inflammasome and keep gut inflammation under control. However, the loss of CCDC50 causes dysregulation of the NLRP3 inflammasome and elevated inflammation and promotes the progression of inflammatory colitis.
These findings that CCDC50-mediated autophagy regulates RLRs-, CGAS-STING1-, and NLRP3-controlled immune and inflammatory signaling pathways are meaningful in human diseases. Some questions remain to be solved. We find that the cellular stress signals can induce CCDC50 expression, but how CCDC50 is activated and regulated by the stimuli and how CCDC50 is recruited to the substrates need further investigation. The signaling hub that controls CCDC50 in autoinflammatory diseases also needs to be revealed.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Reference
- Lin Y, Li Z, Wang Y, et al. CCDC50 suppresses NLRP3 inflammasome activity by mediating autophagic degradation of NLRP3. EMBO Rep. 2022;23:e54453.