
ABSTRACT
In January 2016, the first Epigenetic and Chromatin Regulation of Plant Traits conference was held in Strasbourg, France. An all-star lineup of speakers, a packed audience of 130 participants from over 20 countries, and a friendly scientific atmosphere contributed to make this conference a meeting to remember. In this article we summarize some of the new insights into chromatin, epigenetics, and epigenomics research and highlight nascent ideas and emerging concepts in this exciting area of research.
Introduction
In January 2016, the first Epigenetic and Chromatin Regulation of Plant Traits conference took place in Strasbourg, France, under the joint initiative of 2 EU FP7 Marie Curie Initial Training Network, the CHIP-ET (http://www.chip-et.eu/) and the EpiTRAITS (http://www.epitraits.eu/). With an emphasis on the response to environmental and developmental cues, the 2-day conference provided a comprehensive and up-to-date overview of several aspects of genome regulation by chromatin-mediated and epigenetic mechanisms. Different levels of regulation were highlighted using both experimental and computational approaches, from the DNA/RNA/protein molecular to the chromosomal interaction and nuclear organizational level. New histone variants, new histone modifications, and new proteins were identified and involved in the control of different processes including chromatin structure, transgenerational epigenetic inheritance, and epigenetic somatic memory. Oral presentations from established leaders in the field, emerging researchers, postdoctoral researchers, and PhD students provided the most up-to-date information covering topics related to the regulation of plant traits and the generic nature of chromatin regulation in both plant and non-plant model organisms. In addition, the conference included shorter talks selected from the many excellent abstracts submitted, as well as a lively poster session to stimulate discussions, inspire exchange and reveal mutual interests for putting forward prospects for future experiments and collaborations between research groups from all around the world. In this summary, we report some of the topics discussed during the conference and highlight new insights emerging and future challenges in this growing research area.
Microscopy, molecules, and modeling: Elucidating nuclear organization
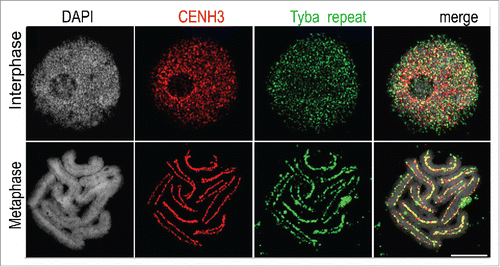
Histones as major components of chromatin are fundamental to genome packaging, function, and regulation. Recent advances connect the microscopically observable organization of the genome to specific histone/chromatin features at the molecular level. Andreas Houben (IPK, Gatersleben, Germany) demonstrated that the sedge Rhynchosporapubera is a plant with holocentric chromosomes (). This phenomenon occurs widely in the monocotyledon families of Cyperaceae and Juncaceae. Holocentric chromosomes possess multiple centromeric units distributed over their entire length. These do not form distinct chromocenters but remain dispersed throughout interphase. For the first time, the underlying centromeric sequences for a holocentric species were identified. In R. pubera, the satellite DNA element termed Tyba and mobile elements interact with the centromeric CENH3-containing nucleosomes. During mitosis CENH3-containing nucleosomes join, forming a polycentromeric band along the chromosomes.Citation1
Figure 1. Holocentromeres in Rhynchospora pubera. Colocalization of the Rhynchospora centromeric histone H3 variant CENH3 (RpCENH3) and the satDNA repeat Tyba on R. pubera holocentromeres. Scale bars: 5 µm (Courtesy of Andreas HoubenCitation1).
A fraction of histones are non-allelic variants whose incorporation in place of canonical histones can lead to dynamic changes in chromatin structure and functions. Frédéric Berger (GMI, Vienna, Austria) presented the work of his group on the histone variant H2A.W in Arabidopsis. This variant is plant-specific and strongly correlated with heterochromatin. Compared to regular nucleosomes, H2A.W containing nucleosomes are wrapped with longer stretches of DNA, enabling long-range chromatin interactions resulting in condensed chromatin fibers. Supporting a role of H2A.W in maintaining condensed chromocenters, mutant plants show dispersed chromocenters. Conversely, the ectopic expression of H2A.W in the central cell of the female gametophyte, which usually does not possess chromocenters, induces chromocenter formation. Finally, while H2A.W acts synergistically with histone H3 dimethylation on lysine 9 (H3K9me2) and DNA methylation in silencing of transposable elements and condensation of heterochromatin, its deposition is independent of these 2 epigenetic marks.Citation2
In Arabidopsis, like in other eukaryotes, the spatial organization of chromatin inside the cell nucleus can be dynamically subject to extensive remodeling throughout development and in response to environmental cues. Fredy Barneche (ENS, Paris, France) gave a report on how light signaling controls nuclear architecture reorganization and chromatin state changes during seedling development. Light exposure induces the rapid formation of chromocenters in cotyledon nuclei after germination and this does not detectably rely on DNA methylation-based processes. This phenomenon is further accompanied by increases endoreduplication and transcriptional activity. Together, these changes are dependent on the light quality and mediated by cryptochromes and the ubiquitin-dependent degradation of the transcription factor (TF) ELONGATED HYPOCOTYL 5 (HY5).Citation3
Another driving force behind elucidating the nuclear architecture is the desire to understand how the organization of chromosomes within the nuclei can affect gene expression. Pawel Mikulski, a PhD student in Daniel Schubert's lab (Free University Berlin, Germany), addressed this question by focusing on the silencing of euchromatic genes. PWWP-DOMAIN INTERACTOR OF POLYCOMBS 1 (PWO1) was identified as a new component of the Polycomb protein pathway required for H3K27me3 on a subset of Polycomb Repressive Complex 2 (PRC2) targets. Interestingly, in addition to interacting with all 3 Arabidopsis PRC2 histone methyltransferases, MEDEA (MEA), CURLY LEAF (CLF), and SWINGER (SWN), PWO1 physically interacts with components of the nuclear membrane, such as CROWDED NUCLEI 1 (CRWN1) or SUN1. Moreover, mutations in PWO1 phenocopy crwn1 plants, resulting in a similar set of genes differentially expressed. Together, it is tempting to imagine that PWO1 may serve as a putative link between Polycomb-dependent silencing of genes and their subnuclear positioning in plants. The question of the impact of the chromosome/nuclear organization on gene expression was also raised in 2 posters. One from Javier Arpon, a PhD student in Philippe Andrey's lab (INRA Versailles, France), focused on the development of statistics and spatial models in order to decipher principles governing the nuclear and genome organization in 3D. The second, from Mariamawit Ashenafi, a PhD student in Celia Baroux's lab (University of Zürich, Switzerland), in which fluorescence in situ hybridization (FISH) was employed to visualize the localization of active versus silenced genes relative to heterochromatin and the nuclear periphery.
The involvement of the interface between the nuclear envelope and the nucleoplasm on the nuclear organization was further addressed in 2 related posters. Christophe Tatout (Clermont University, France) presented a poster focused on the role of SUN domain proteins (known as key components of the linker of the nucleoskeleton and cytoskeleton complex) on the organization of heterochromatin in Arabidopsis interphase nuclei. It was reported that the nuclear shape is altered and the silencing of heterochromatic repeats is alleviated in sun mutants. Related to this, Marie-Edith Chabouté (IBMP, Strasbourg, France) presented a poster about the functional characterization of the newly found γ–tubulin associated proteins GCP3-INTERACTING PROTEIN 1 and 2 (GIP1 and GIP2), which colocalized with the centromere-specific H3 variant CENH3 and contributed to the regulation of centromere at the interface between the nuclear envelope and the nucleoplasm.Citation4,5
While this was a conference for plant epigenetics, several invited speakers gave us the opportunity to expose our minds to other organisms. Wendy Bickmore (Institute of Genetics and Molecular Medicine, Edinburgh) discussed the relationships between structure and function in human cell nuclei, with the crucial issue of whether and to what extent the nuclear organization can directly affect gene function rather than merely reflecting it. Using the Escherichia coli lacO/lacI tethering system in human cells, Wendy Bickmore's group had observed that the artificial relocation of some human genes close to the nuclear periphery could lead to the reduction of their expression, while the expression of many neighboring genes remains unchanged. This work suggests that the relocalization of a gene relative to the nuclear periphery can be used to modulate its expression during development and differentiation without necessarily altering expression of their neighbor genes.Citation6 Next, she addressed the question whether changes in transcription can impose changes in spatial organization? Using synthetic transcriptional activators (TALEs), the Bickmore's group also found that nuclear reorganization is driven by chromatin remodeling rather than transcription.Citation7
Finally, Dieter Heermann (Heidelberg University, Germany) offered a different approach to spatial chromatin organization by polymer modeling.Citation8 In a prokaryotic cell with a circular chromosome, the confined space alone already leads to supercoiling and looping. Adding single DNA interacting proteins into the model can explain spatial segregation between 2 chromosomes after replication. Moreover, chromatin regions are predicted to disentangle spontaneously if different looping parameters are established by sequence specific binding of proteins. This spatial segregation leads to the formation of compartments and enables the chemical separation of molecules.
Dare to be a force of change: Chromatin remodeling during plant development
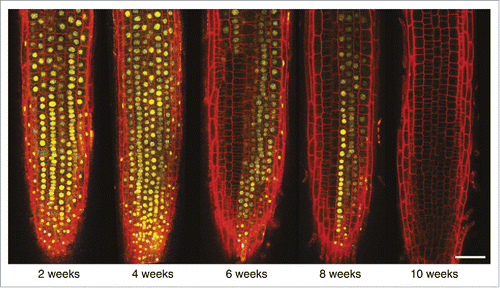
In eukaryotes, dynamic changes in chromatin states are known to play central roles in establishing particular expression patterns during development and can be further maintained within a cell lineage through mitosis in order to remember past events. In plants, vernalization is a classic epigenetic process in which prolonged cold exposure quantitatively affects the timing of the floral transition. During this process, the Polycomb target gene and floral repressor FLOWERING LOCUS C (FLC) is epigenetically repressed, independently of cytosine methylation, through a local increase in the repressive mark H3K27me3.Citation9 The length of the cold period is epigenetically remembered throughout plant development until embryogenesis, where reactivation of FLC resets the vernalization requirement for the next generation.Citation10 During his talk, Martin Howard (John Innes Center, United Kingdom) addressed the question of the mechanistic basis of this quantitative epigenetic memory. Using mathematical modeling coupled with experiments such as fluorescent imaging, his group discovered that the epigenetic memory of the FLC system is stored locally in its chromatin, for example, in the form of histone modifications (cis memory), and not in networks of diffusible factors (trans memory; ).Citation11 Also focusing on FLC and using mathematical modeling and experiments, Martin presented evidence that both the nature of temperature registration at FLC and its cis-based epigenetic memory are both digital.Citation11,12 The question now is how this digital epigenetic regulation and the more fine-tuned analog transcriptional control can be combined to provide plants with a robust yet flexible developmental program.
The regulation of flowering was also addressed through various chromatin-related processes in several posters. In Arabidopsis, the acetylation of histone H4 by the NuA4 complex was previously proposed to participate in the transcriptional regulation of FLC.Citation13 Alfonso Mouriz, from Manuel Piñeiro's group (INIA-UPM Madrid, Spain), presented the characterization of several NuA4 subunits involved in this process, with the different flowering phenotypes of their corresponding loss-of-function mutants. From the same group, Dorota Komar (INIA-UPM Madrid, Spain) analyzed the role of EARLY BOLTING IN SHORT DAYS (EBS)Citation14 in controlling the expression of multiple flowering time genes. Using a genetic approach, Wei Zhao, a PhD student in Wen-Hui Shen's group (IBMP, Strasbourg, France) described his work on a double mutant defective in both H3K36 methylation and H2B monoubiquitination. While on a global level these histone marks seem largely independent, his results suggested a possible crosstalk between them at some flowering time genes. Then, bringing together gene regulation and spatial nuclear organization, Stefania Del Prete, a PhD student in Valérie Gaudin's group (INRA Versailles, France) reported in her poster how by using Arabidopsis transgenic lines she conditionally modulates LHP1 expression to follow chromatin events and nuclear organization changes that accompany the floral transition.
Connected with flowering and using the Arabidopsis floral meristem as a model, Cristel Carles (Grenoble Alpes University, France) presented the ongoing work of her group on the study of how chromatin changes can drive cell fate specification in Arabidopsis. For this purpose, the role of ULTRAPETALA1 (UTL1) in controlling shoot and floral meristem activities by targeting key flower developmental genes was addressed.Citation15 On one hand, ULT1 functions antagonistically with CLF preventing CLF from methylating H3K27, likely through their protein-protein interaction and thus de-repressing target developmental genes. On the other hand, ULT1 interacts with the histone methyltransferase ARABIDOPSIS HOMOLOG OF TRITHORAX1 (ATX1) and the RNA polymerase (RNAP) II C-terminal domain and promotes deposition of the active mark H3K4me3 and transcription initiation.Citation16 ULT1 thus seems to function as a linker between chromatin remodeling and transcriptional regulation. Another aspect of Cristel Carles's work is devoted to the understanding of how chromatin is brought from repressed to active at developmental loci. Using large-scale approaches in a floral induction system,Citation17 her group demonstrated that changes in the active H3K4me3 and the repressive H3K27me3 histone marks are highly correlated with expression changes. Providing us with a dynamic view of chromatin changes throughout floral development, Carles further reported that early events of gene activation are dominated by an increase in H3K4me3, while a strong decrease in H3K27me3 occurs later.
Among other mechanisms, DNA methylation is involved in many aspects of chromatin function, including gene expression, silencing, and DNA repair. In this respect, Philippe Gallusci (Bordeaux University, France) gave an overview of his team's work on DNA methylation dynamics during fruit ripening, an important developmental process unique to plants. Gallusci demonstrated that the global cytosine methylation level was decreased by 30% in pericarp of tomato fruits during ripening (). As DNA replication is very limited at this stage, which excludes the idea of a DNA methylation “dilution” through replication, this decrease was connected with a previously unknown active DNA demethylation process.Citation18 Indeed, they later on identified the molecular mechanism of this active DNA demethylation as being mainly mediated by the tomato DEMETER-like DNA demethylase SlDML2.Citation19 This finding is especially interesting when considering the timing and extent of DNA demethylation as a probable source of variation in the diversity of kinetics and intensity of ripening among tomato varieties. Moreover, because changes in DNA methylation were also reported during pear and apple ripening,Citation20,21 one can ask whether such mechanism is conserved in all fleshy fruit species.
Figure 2. The digital mechanism of FLC repression in response to vernalization. FLC-Venus regulation in the root meristem follow a binary On/Off states being either fully expressed or fully repressed in the single cell. The transcriptional state is transmitted to the daughter cells through mitotic divisions. Plants were subjected to 2, 4, 6, 8, or 10 weeks of cold exposure and imaged 7 d after return to warm by confocal microscopy. Scale bar = 50 μm (Courtesy of Martin Howard).
Genomic imprinting is an epigenetic phenomenon involving both histone modifications and DNA methylation that has independently evolved in flowering plants and mammals. This phenomenon corresponds to the differential expression of genetically identical alleles based on their parent-of-origin. In plants, imprinting is largely confined to the embryo-nourishing endosperm tissue. Claudia Köhler (Uppsala, Sweden) gave an overview of her research on genomic imprinting establishment and evolution (). Combining the Isolation of Nuclei TAgged in specific Cell Types (INTACT) method together with epigenome profiling, she showed that the H3K27me3 distribution is strikingly different in leaves and endosperm. While H3K27me3 is enriched in pericentromeric regions in the endosperm, it is excluded from pericentromeric regions in leaf tissues. Also, maternal-specific H3K27me3 regions are distributed throughout the genome, while paternal-specific ones are more confined to pericentromeric regions. Interestingly, the allele-specific H3K27me3 localization to the maternal allele of paternally expressed genes is targeted to DNA hypomethylated sites, while, in the paternal genome, H3K27me3 is enriched at transposable elements (TEs) located in pericentromeric heterochromatin (i.e., Gypsy) and is negatively correlated with the heterochromatic mark H3K9me2. Finally, comparing imprinted genes between Arabidopsis and its close relative Capsella rubella, covering an evolutionary period of around 10-14 million years,Citation22 it was proposed that imprinted maternally expressed genes are not regulated by H3K27me3 but instead by the RNA-directed DNA methylation (RdDM) pathway through trans-acting sRNAs. Together, it can be concluded that genomic imprinting is a rapidly evolving process arising from the epigenetic regulation of TEs.
Figure 3. Active DNA demethylation controls tomato fruit ripening. While the breaker stage (Br) occurs around 39 d postanthesis (dpa) in wild type tomato fruits, ripening is strongly inhibited in an RNAi DML knockdown line. Scale bars: 1 cm. (Courtesy of Ruie Liu and Philippe Gallusci).

Chromatin alteration: Cause or consequence of the response to environmental cues
Plants are sessile organisms; therefore, they have to get along with the many stresses and fast transient changes of their growing conditions during their lifetime. Fortunately, plants possess an enormous developmental plasticity, which plays a crucial role for stress response and adaptation. Ambient temperature is a major determinant of plant development and effective growth and small temperature changes can influence the timing of flowering. In Arabidopsis Col-0 and Ler accessions, while a high ambient temperature (25°C) will accelerate flowering, a low one (16°C) will, conversely, delay flowering.Citation23 Previous studies have shown that the MADS-box TF genes FLOWERING LOCUS M (FLM), MADS AFFECTING FLOWERING 2 (MAF2) and SHORT VEGETATIVE PHASE (SVP) play key roles in the temperature-dependent regulation of flowering, with the alternative splicing of FLM and MAF2 working as a molecular thermometer to prevent or induce precocious flowering.Citation24,25 Alice Pajoro, post-doc in Richard Immink's group (Wageningen University, The Netherlands), reported her work on the epigenetic regulation behind this temperature-mediated alternative splicing process. Using RNA-seq, numerous temperature-mediated alternative splicing events, including many flowering time regulators, were identified. At the epigenome level, she showed preliminary results suggesting that the level of H3K36me3 rises upon increasing ambient temperature. Related to this exciting finding, mutants affected in H3K36me3, such as sdg8 or sdg26, were reported to flower at the same time independently of the experienced ambient temperature.Citation26 Therefore, a question remains open as to the effect of the lack of H3K36me3 on temperature-mediated alternative splicing events.
Another mechanism used to coordinate the ambient temperature transcriptome is based on the deposition/removal of the histone variant H2A.Z into nucleosomes in place of H2A by the chromatin remodeling factor ACTIN-RELATED PROTEIN 6 (ARP6). Under warm temperature, H2A.Z is evicted especially near the transcriptional start site of the heat-inducible heat shock protein 70 (HSP70) gene at the +1 nucleosome. This influences the ability of RNAP II to transcribe this gene, thus eliciting a temperature response.Citation27 In agreement with this scenario, the arp6 mutant, which is deficient in H2A.Z deposition, displays, among other phenotypes, a constitutive warm temperature response. Sandra Cortijo, from Phil Wigge's group (The Sainsbury Laboratory, Cambridge, UK), presented new insight into the interplay between the H2A.Z occupancy and transcriptional changes in response to temperature variations. Through large-scale approaches, correlations between the temperature transcriptome, the nucleosome density, and the H2A.Z-nucleosome occupancy were dynamically addressed in response to a sudden temperature increase (from 17°C to 27°C). Following this strategy, a fast, targeted, and transient eviction of H2A.Z at the +1 nucleosome was detected for a hundred genes activated by the temperature shift. Moreover, a strong correlation was observed between the binding of the HEAT SHOCK FACTOR PROTEIN 1 along the entire transcript unit and the H2A.Z eviction at the +1 nucleosome at temperature activated genes.
Also addressing the central contribution of epigenetics to stress adaptation, Ortrun Mittelsten Scheid (GMI, Vienna, Austria) gave an integrated view of the dynamic changes in chromatin, nuclear organization, and transcription during the implementation of plant stress responses in Arabidopsis.Citation28 While the response to acute heat stress is fast and induces heat shock TFs, additional sequences are transcriptionally activated in case of prolonged heat stress: TEs, such as the multicopy ONSEN retrotransposon, and further repeat families, such as the pericentromeric TSI repeat.Citation29 This is accompanied by a transient reduction in nucleosome density. Upon recovery from heat stress, nucleosome loading and transcriptional silencing are overcompensated and finally restored, including heterochromatin condensation. While addressing the question of the transgenerational transmission of heat stress-related information, no heritable effects were observed so far. Together, the 3 works summarized here perfectly illustrate the very complex and dynamic interplay that exists between transcription, RNA splicing, and chromatin remodeling in order to fine-tune the plant response to temperature variations.
Beside nucleosomal histones, H1 linker histones are also essential for the maintenance of the chromatin structure and for the regulation of gene expression in higher eukaryotes. Following this statement, Andrzej Jerzmanowski (Warsaw University and Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Poland) gave a report about the crucial role played by the stress-inducible H1.3 variant in the adaptive responses to environmental changes. Under normal growth conditions, H1.3 is detectable almost exclusively in guard cells where it acts as a regulator of stomatal functions, while under combined light and water deficiency, its expression undergoes a strong and systemic induction. Supporting H1.3 as the most dynamic variant, this increase was found to be only transient after restoring standard growth conditions. Moreover, H1.3 has no stably bound pool in the nucleus and is exchanged in chromatin extremely rapidly. Genome-wide distribution of the 3 H1 non-allelic variants in Arabidopsis plants grown in low light or in control conditions by ChIP-chip revealed some similarities and discrepancies between the occupancy profiles of H1.3 and the main H1 variants. While all the variants bind predominantly heterochromatic regions, H1.3 has greater tendency to associate with chromatin enriched in marks of active transcription (H3K4me3). The analysis of genome-wide cytosine methylation patterns further revealed relation between H1.3 and global stress-related DNA hypermethylation. In contrast, H1.1 and H1.2 were previously found to prevent the access of DNA methyltransferases to DNA.Citation30 Thus, this work supports the crucial role played by H1.3 in facilitating chromatin accessibility in response to particular environmental signals.Citation31
Epigenetic processes and changes in response to drought stress were also approached during this conference. Barbara Correia, supervised by Glória Pinto (University of Aveiro, Portugal), showed how she followed the severity of drought stress and relief by quantifying several biochemical markers of oxidative stress and DNA methylation patterns in Eucalyptus globulus leaves. The global cytosine methylation was found increased over the period of dehydration, especially in the vascular tissue, but it rapidly decreased after rehydration. Korana Surdonja, a PhD student in Markus Kuhlmann's group (IPK Gatersleben, Germany), presented an analysis of short interfering RNA (siRNA) populations after drought stress in barley seeds. Despite the general reduction in siRNAs, stress-specific 24-mers and their putative targets were identified and related to DNA methylation and transcriptional changes. Among others, the cytokinin oxidase CKX showed changes in DNA methylation in its promoter in the stress treated seeds. Finally, Aurine Verkest (Bayer Crop Science, Ghent, Belgium) reported about her recently published work on transcriptome and histone changes in canola isogenic epi-lines selected for increased energy use efficiency and drought tolerance.Citation32 For instance, differential H3K4me3 distribution was identified targeting drought responsive genes, confirming the effectivity of the epi-selection.
“In movement:” Chromatin remodeling research stays in shape
In addition to being essential for gene transcription, chromatin remodeling also participates in other DNA-template processes. A number of speakers gave insights about chromatin remodeling and its involvement in processes such as replication, homologous recombination (HR), and DNA repair. In order to expand our knowledge of posttranslational histone modifications in plants, Minerva Trejo-Arellano, from Lars Hennig's group (Uppsala, Sweden), presented the discovery of a new heterochromatic histone mark in Arabidopsis, H3K23me1. This mark was identified by mass spectrometry and its genome-wide distribution was further analyzed using ChIP-seq. In this way, H3K23me1 was found enriched in pericentromeric regions, mostly in heterochromatic repetitive DNA sequences and TEs. H3K23me1 did not correlate with gene expression but was associated with the heterochromatic mark H3K9me2. Moreover, the histone methyltransferase KYP/SUVH4, which deposits H3K9 methylation, was also required for full H3K23me1 levels. Finally, H3K23me1 was found to colocalize with DNA methylation in the 3 cytosine methylation contexts at TEs.
Focusing on another aspect of chromatin remodeling, Crisanto Gutierrez (CBM, Madrid, Spain) reported about a recent goal of his group to correlate regulation of S-phase during cell cycle with chromatin organization. Using BrdU-labeling on synchronized cell cultures, the group identified locations of DNA replication origins (ORIs) along Arabidopsis chromosomes.Citation33 Comparison of ORI location with the chromatin landscape revealed a negative correlation between ORIs and DNA methylation and a positive one between ORIs and canonical marks of active chromatin, as observed in mammalian cells.Citation34,35 Interestingly, while most ORIs preferentially colocalize with genes, around 10% of them are present in heterochromatin and associate with particular classes of TE in both non-pericentromeric and pericentromeric regions. A major challenge ahead will be to identify ORIs in the whole plant, a project that is being pursued using Short Nascent Strands (SNS)-seq purified from seedlings. Preliminary results from SNS-seq, combined with the epigenomic data on chromatin organization,Citation36,37 showed a predominant enrichment of ORI location in certain chromatin states. Next, it will be exciting to decipher the precise causal relationship between chromatin modulation and DNA replication control.
The involvement of chromatin remodeling in DNA repair was highlighted by Wen-Hui Shen (IBMP, Strasbourg, France). Using GUS recombination reporter systems developed by the group of Barbara HohnCitation38 (present in the audience), Wen-Hui Shen's group proved that the NUCLEOSOME ASSEMBLY PROTEIN1 (NAP1) and NAP1-RELATED PROTEIN (NRP) families of H2A/B histone chaperones are required for somatic HR in Arabidopsis.Citation39 Further experiments revealed that proteins of the NAP1 and NRP groups act synergistically in the somatic HR pathway, resulting in the increased sensitivity of their mutants to genotoxic or abiotic stresses. Moreover, in agreement with known impact of chromatin remodelers on genome stability,Citation40 a connection between the NAP1/NRP groups and INO80 was presented. Together, these data provide new insight into our understanding of the distinct functions of different families of epigenetic regulators in DNA repair processes and genome protection within the chromatin context.
Looking at chromatin dynamics evolution on a wide time-scale, François Roudier (ENS, Paris, France) presented results from a cross-species epigenomic analysis done on 3 Brassicaceae representatives: Arabidopsis thaliana, Arabidopsis lyrata, and Arabis alpina, covering a time frame from 5 to 40 million years.Citation41 The comparison of the 3 corresponding epigenomes revealed an overall conservation of H3K4me3 and H3K27me3 distribution, with some lineage-specific variations. In particular, 2 types of H3K27me3 domains, conserved and dynamic over the last 40 million years, were identified. Distinction between these 2 types was reflected by differential promoter sequence information content, nucleosome occupancy, as well as functional classification and expression pattern of H3K27me3-marked genes. In addition, syntenic blocks enriched in conserved (ancestrally-derived) H3K27me3 domains showed their preferential association with genomic regions involved in many long-range intra-chromosomal interactions that have been reported elsewhere.Citation42,43 These outcomes point out the influence of multiple evolutionary constraints on PRC2-mediated gene repression and the existence of distinct sets of H3K27me3-marked regions, thus explaining the difficulty to identify cis-regulatory elements common to all PRC2 targets.
Enhancer quest: Catch me if you can
Enhancers are cis-regulatory elements able to activate genes from relatively long distance (up to megabases). Enhancers are involved in fine-tuning transcription of genes and are key regulators of cell differentiation and development. Despite their central role, enhancers are difficult to identify accurately at a genome-wide scale. Albin Sandelin (University of Copenhagen, Denmark) presented evidence showing differences of transcriptional timing between enhancers and promoters in a wide range of human and mouse cell types and in response to different stimuli.Citation44 Exploiting the fact that many enhancers are transcribed into enhancer RNA when active, Albin Sandelin's group used 5′ end sequencing (CAGE) to profile enhancer activation. Through this approach, they found that enhancer transcription is preceding gene promoter transcription, challenging the previous idea that enhancers and promoters were co-expressed.Citation45 Therefore, upon cell differentiation, enhancers were transcribed first, followed by genes coding for TFs, and finally by genes coding for proteins other than TFs. All together, these findings bring a global view of the complex and dynamic transcriptional changes occurring during cell differentiation in mammals.
Blaise Weber, from Maike Stam's group (University of Amsterdam, the Netherlands), presented results of a collaborative project with Franziska Turck's group (Max Planck Institute for Plant Breeding Research, Cologne, Germany) on enhancer prediction using newly generated genome-wide data in maize. In animals, active enhancers were found to be associated with nucleosome-depleted regions (NDRs) and specific histone marks, such as H3K27ac and H3K9ac.Citation46–48 In this work, maps of NDRs (by DNase-seq) and H3K9ac (by ChIP-seq) were generated in maize using 2 different tissues (young seedling leaves and husk). In addition, RNA-seq was performed to assess differences in gene expression levels between these 2 tissues. The combination of these 3 large-scale approaches was then used to identify putative enhancers. A list of around 3,000 candidate enhancers was obtained. Differential presence of the chromatin features tested allowed distinction between tissue-specific and constitutive enhancer candidates. A subset of the enhancer candidates is currently being validated using transgenic reporter assays.
Maike Stam, group leader at the University of Amsterdam (the Netherlands), presented their research on the essential role of MEDIATOR OF PARAMUTATION 1 (MOP1) in paramutation and enhancer activity. Paramutation is defined as a trans-interaction between 2 alleles of the same gene that leads to the epigenetic silencing of one allele.Citation49,50 The RdDM machinery is shown to be required for paramutation. One of the hallmarks of this pathway is DNA methylation in the CHH context. In maize, the b1 locus, coding for a TF involved in the anthocyanin pathway, exhibits 2 epiallelic states whereby b1 is either lowly transcribed (B') or highly transcribed (B-I). A hundred kilobases upstream from the gene, multiple tandem repeats act as long-distance enhancer and are marked with differing epigenetic modifications at the 2 epialleles. These repeats are essential for paramutation to happen Furthermore, using targeted bisulfite sequencing and genome-wide analysis, the group of Maike Stam demonstrated that RdDM loci can also be associated with low CHH methylation.
Advancing the comprehension of epigenetics and heritability
In addition to the Mendelian heredity of polymorphic DNA sequences, epigenetic changes can also modify gene expression in a heritable manner. In plants, cytosine methylation for transcriptional gene silencing can occur through RdDM driven by DNA-dependent RNAP IV and V Citation51. This RdDM is lost in pol IV or pol V null mutants, but is restored upon genetic complementation or outcrossing with the wild type, suggesting the existence of a silent locus identity propagated independently of RNAP IV/V-dependent RdDM. Todd Blevins, now group leader at the IBMP (Strasbourg, France), presented results from his post-doc in Craig Pikaard's group. Blevins and colleagues found that the maintenance of silent locus identity and production of 24 nt siRNAs, which specify targets of RdDM, requires HISTONE DEACETYLASE 6 (HDA6) and the DMNT1-like cytosine methyltransferase MET1 at certain loci.Citation52 Maintaining this silent locus identity and gene silencing via RdDM are 2 separable steps that permit the epigenetic inheritance of a silent state. Loss of HDA6 function gives rise to reactivated epialleles. Using combinations of Arabidopsis ecotypes, Todd further demonstrated a rescue of lethality and genetic hybrid incompatibility via reversion of epiallele silencing. Using the example of a duplicated essential gene pair, for which differential cytosine methylation and expression result in lethality in hybrid progeny, Blevins showed that this hybrid incompatibility can be rescued in an hda6 mutant background due to the reactivation of a previously silent gene paralog.
Stability of DNA methylation was also the focus of a poster presented by Johan Zicola, a Ph.D student in the group of Franziska Turck (Max Planck Institute for Plant Breeding Research in Cologne, Germany). Zicola showed data from Arabidopsis lines in which DNA methylation was induced at an enhancer of the FLOWERING LOCUS T (FT) gene using an inverted-repeat. This resulted in the downregulation of FT expression and a stably delayed flowering phenotype across generations. Thus, inverted-repeats might be used as an interesting tool to characterize enhancers and fine-tune gene expression.
Epigenetic changes and their heritability are correlated with agronomic performance. Energy efficiency and energy homeostasis have an epigenetic component that can be directed and stabilized by using artificial selection, resulting in superior agronomic traits in crops.Citation53,54 For example, Brassica napus isogenic lines artificially selected for their enhanced drought stress tolerance displayed upregulated transcriptomic stress response related with particular epigenetic variations, including differential H3K4me3 at several drought-responsive genes.Citation32 Martin Schmidt, from Mieke Van Lijsebettens's group (VIB, PSB, Ghent University, Ghent, Belgium), presented work on a selected Oryza sativa line with improved energy use efficiency and seed yield. By focusing on the stability of particular epigenetic variations and in order to identify candidate epialleles for those traits, cytosine methylation changes were analyzed by genome-wide bisulfite sequencing and correlated with differential gene transcription analyzed by RNA-seq.
TEs account for a significant proportion of eukaryotic genomes and their movement/accumulation are considered to be major forces shaping plant genomes.Citation55 To tame TE activity, plant genomes have elaborate epigenetic strategies from which TE can escape under certain conditions. Despite this essential role, studying TE activity as a mobile part of the genome (referred as the mobilome) is still very challenging. Sophie Lanciano, supervised by Marie Mirouze (IRD, Perpignan, France), presented a new method to detect active retrotransposons by high-throughput sequencing of isolated extrachromosomal circular DNA. The proof of concept was carried out in Arabidopsis and rice samples with destabilized epigenomes. Using this very exciting method, known active retrotransposons were further confirmed (e.g., Tos17 in rice) and new ongoing retrotransposons activities were identified during seed development.
Heterosis classically referred to the improved vigor, including greater biomass, rapid development, and increased fertility, of F1 hybrids in comparison with their parental homozygous inbred lines. This phenomenon is a fundamental issue in plant breeding and several posters were focused on its epigenetic features. Vania Helena Techio (Federal University of Lavras, Brazil) reported about the chromosomal distribution of histone modifications and DNA methylation in interspecific hybrids of Brachiaria. A correlation between H3K9me2 and cytosine methylation was observed in nucleolus organizer regions, centromeric central domains and pericentromeric regions, while H3K4me2 was detected in euchromatic domains, mainly in the terminal chromosomal regions. Asymmetry in DNA methylation between sister chromatids was also observed.Citation56 Similarly, Kathrin Lauss, a PhD student in Maike Stam's group (University of Amsterdam, the Netherlands), described her research on Arabidopsis epiHybrids generated from epigenetic recombinant inbred lines, which differ in their DNA methylation while being nearly isogenic. In this way, numerous examples of positive and negative heterosis in quantitative traits were observed. Susanne Edelmann, from Stefan Scholten's group (University of Hamburg, Germany), presented a poster aiming on the question of whether DNA methylation patterns established during early embryogenesis can impact upon the heterotic response in maize. By using a methyltransferase inhibitor, successful DNA demethylation in early embryos of hybrids and inbred lines was first confirmed and, by comparing seedlings derived from treated embryos to their untreated controls, an increase in growth heterosis was observed. A second poster from Stefan Scholten's group explored the contribution of mobile small RNAs (sRNA) on heterosis. Stefan's group showed the extremely high variability between sRNA populations of maize parental lines of different heterotic groups. An association study revealed a highly significant correlation between yield heterosis and specific sets of 22- and 24-nt sRNA. Strikingly, experiments to reduce sRNA transcriptome complexity increased heterosis, suggesting a direct, overall negative involvement of sRNA in heterosis.
Epigenetic and chromatin, still a flourishing field of research
There is no doubt that epigenetics is a determinant part of the fine-tuning of gene regulation, which conditions the proper response of plants to environmental changes and eventually their evolutionary success. Our current knowledge suggests that histone variants and their posttranslational modifications, cytosine methylation, non-coding RNAs, as well as chromatin rearrangements, are involved in almost every aspect of plant life, including the development of agronomically important traits. However, despite the advances in genomics, proteomics, phenomics, and high-throughput sequencing methodologies, the causal link between changes in gene expression and epigenetic mark deposition is still difficult to establish and remains one of the most challenging questions in the field of epigenetics. Nevertheless, works reported in this conference have made significant headway in solving this chicken or egg dilemma, by proposing new techniques, detailed description of changing chromatin states, and multidisciplinary approaches joining the forces of biology, genetics, and the power of computational strategies.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We warmly thank all participants for sharing their published/unpublished data, for their proactive and exciting discussion during the meeting and especially during the poster session. We apologize to those whose contributions could not be mentioned due to format restrictions. We acknowledge the financial support of the Strasbourg University and its Mission Investissementd'Avenir, the Mission Investissement d'Avenir European Union Seventh Framework Program and the European Commission Marie Curie Research Training Networks ‘Epigenetic regulation of economically important plant traits’ (EpiTRAITS, FP7-PEOPLE-2012-ITN316965) and ‘Chromatin in Plants – European Training and Mobility’ (CHIP-ET, FP7-PEOPLE-2013-ITN607880).
References
- Marques A, Ribeiro T, Neumann P, Macas J, Novák P, Schubert V, Pellino M, Fuchs J, Ma W, Kuhlmann M, et al. Holocentromeres in Rhynchospora are associated with genome-wide centromere-specific repeat arrays interspersed among euchromatin. Proc Natl Acad Sci 2015; 112:13633-8; PMID:26489653; http://dx.doi.org/10.1073/pnas.1512255112
- Yelagandula R, Stroud H, Holec S, Zhou K, Feng S, Zhong X, Muthurajan UM, Nie X, Kawashima T, Groth M, et al. The histone variant H2A.W defines heterochromatin and promotes chromatin condensation in Arabidopsis. Cell 2014; 158:98-109; PMID:24995981; http://dx.doi.org/10.1016/j.cell.2014.06.006
- Bourbousse C, Mestiri I, Zabulon G, Bourge M, Formiggini F, Koini MA, Brown SC, Fransz P, Bowler C, Barneche F. Light signaling controls nuclear architecture reorganization during seedling establishment. Proc Natl Acad Sci 2015; 112:E2836-44; PMID:25964332; http://dx.doi.org/10.1073/pnas.1503512112
- Batzenschlager M, Lermontova I, Schubert V, Fuchs J, Berr A, Koini MA, Houlné G, Herzog E, Rutten T, Alioua A, et al. Arabidopsis MZT1 homologs GIP1 and GIP2 are essential for centromere architecture. Proc Natl Acad Sci U S A 2015; 112:8656-60; PMID:26124146; http://dx.doi.org/10.1073/pnas.1506351112
- Chabouté M-E, Berr A. GIP contributions to the regulation of centromere at the interface between the nuclear envelope and the nucleoplasm. Front Plant Sci 2016; 7:118; PMID:26904080; http://dx.doi.org/10.3389/fpls.2016.00118
- Finlan LE, Sproul D, Thomson I, Boyle S, Kerr E, Perry P, Ylstra B, Chubb JR, Bickmore WA. Recruitment to the nuclear periphery can alter expression of genes in human cells. PLoS Genet 2008; 4:e1000039; PMID:18369458; http://dx.doi.org/10.1371/journal.pgen.1000039
- Therizols P, Illingworth RS, Courilleau C, Boyle S, Wood AJ, Bickmore WA. Chromatin decondensation is sufficient to alter nuclear organization in embryonic stem cells. Science 2014; 346:1238-42; PMID:25477464; http://dx.doi.org/10.1126/science.1259587
- Di Ventura B, Knecht B, Andreas H, Godinez WJ, Fritsche M, Rohr K, Nickel W, Heermann DW, Sourjik V. Chromosome segregation by the Escherichia coli Min system. Mol Syst Biol 2013; 9:686; PMID:24022004; http://dx.doi.org/10.1038/msb.2013.44
- Finnegan JE, Kovac KA, Jaligot E, Sheldon CC, Peacock WJ, Dennis ES. The downregulation of FLOWERING LOCUS C (FLC) expression in plants with low levels of DNA methylation and by vernalization occurs by distinct mechanisms. Plant J Cell Mol Biol 2005; 44:420-32; PMID:16236152; http://dx.doi.org/10.1111/j.1365-313X.2005.02541.x
- Iwasaki M. Chromatin resetting mechanisms preventing trangenerational inheritance of epigenetic states. Front Plant Sci 2015; 6:380; PMID:26074941; http://dx.doi.org/10.3389/fpls.2015.00380
- Berry S, Hartley M, Olsson TSG, Dean C, Howard M. Local chromatin environment of a Polycomb target gene instructs its own epigenetic inheritance. eLife 2015; 4:e07205; PMID:26074941; http://dx.doi.org/10.7554/eLife.07205
- Angel A, Song J, Yang H, Questa JI, Dean C, Howard M. Vernalizing cold is registered digitally at FLC. Proc Natl Acad Sci 2015; 112:4146-51; PMID:25775579; http://dx.doi.org/10.1073/pnas.1503100112
- Zacharaki V, Benhamed M, Poulios S, Latrasse D, Papoutsoglou P, Delarue M, Vlachonasios KE. The Arabidopsis ortholog of the YEATS domain containing protein YAF9a regulates flowering by controlling H4 acetylation levels at the FLC locus. Plant Sci Int J Exp Plant Biol 2012; 196:44-52; PMID:23017898 http://dx.doi.org/10.1016/j.plantsci.2012.07.010
- López-González L, Mouriz A, Narro-Diego L, Bustos R, Martínez-Zapater JM, Jarillo JA, Piñeiro M. Chromatin-dependent repression of the arabidopsis floral integrator genes involves plant specific PHD-Containing proteins. Plant Cell Online 2014; 26:3922-38; PMID:25281686 http://dx.doi.org/10.1105/tpc.114.130781
- Moreau F, Thévenon E, Blanvillain R, Lopez-Vidriero I, Franco-Zorrilla JM, Dumas R, Parcy F, Morel P, Trehin C, Carles CC. The Myb-domain protein ULTRAPETALA1 INTERACTING FACTOR 1 controls floral meristem activities in Arabidopsis. Dev Camb Engl 2016; 143:1108-19; 26903506 http://dx.doi.org/10.1242/dev.127365
- Engelhorn J, Moreau F, Fletcher JC, Carles CC. ULTRAPETALA1 and LEAFY pathways function independently in specifying identity and determinacy at the Arabidopsis floral meristem. Ann Bot 2014; 114:1497-505; PMID:25288633; http://dx.doi.org/10.1093/aob/mcu185
- Wellmer F, Alves-Ferreira M, Dubois A, Riechmann JL, Meyerowitz EM. Genome-Wide Analysis of Gene Expression during Early Arabidopsis Flower Development. PLoS Genet 2006; 2:e117; PMID:16789830; http://dx.doi.org/10.1371/journal.pgen.0020117
- Teyssier E, Bernacchia G, Maury S, Kit AH, Stammitti-Bert L, Rolin D, Gallusci P. Tissue dependent variations of DNA methylation and endoreduplication levels during tomato fruit development and ripening. Planta 2008; 228:391-9; PMID:18488247; http://dx.doi.org/10.1007/s00425-008-0743-z
- Liu R, How-Kit A, Stammitti L, Teyssier E, Rolin D, Mortain-Bertrand A, Halle S, Liu M, Kong J, Wu C, et al. A DEMETER-like DNA demethylase governs tomato fruit ripening. Proc Natl Acad Sci 2015; 112:10804-9; PMID:26261318; http://dx.doi.org/10.1073/pnas.1503362112
- Telias A, Lin-Wang K, Stevenson DE, Cooney JM, Hellens RP, Allan AC, Hoover EE, Bradeen JM. Apple skin patterning is associated with differential expression of MYB10. BMC Plant Biol 2011; 11:93; PMID:21599973; http://dx.doi.org/10.1186/1471-2229-11-93
- Wang Z, Meng D, Wang A, Li T, Jiang S, Cong P, Li T. The Methylation of the PcMYB10 Promoter Is Associated with Green-Skinned Sport in Max Red Bartlett Pear. PLANT Physiol 2013; 162:885-96; PMID:23629835; http://dx.doi.org/10.1104/pp.113.214700
- Koch MA, Kiefer M. Genome evolution among cruciferous plants: a lecture from the comparison of the genetic maps of three diploid species—Capsella rubella, Arabidopsis lyrata subsp. petraea, and A. thaliana. Am J Bot 2005; 92:761-7; PMID:21652456; http://dx.doi.org/10.3732/ajb.92.4.761
- Balasubramanian S, Sureshkumar S, Lempe J, Weigel D. Potent Induction of Arabidopsis thaliana Flowering by Elevated Growth Temperature. PLoS Genet 2006; 2:e106; PMID:16839183; http://dx.doi.org/10.1371/journal.pgen.0020106
- Pose D, Verhage L, Ott F, Yant L, Mathieu J, Angenent GC, Immink RGH, Schmid M. Temperature-dependent regulation of flowering by antagonistic FLM variants. Nature 2013; 503:414-7; PMID:24067612; http://dx.doi.org/10.1038/nature12633
- Airoldi CA, McKay M, Davies B. MAF2 Is Regulated by Temperature-Dependent Splicing and Represses Flowering at Low Temperatures in Parallel with FLM. PLoS ONE 2015; 10:e0126516; PMID:25955034; http://dx.doi.org/10.1371/journal.pone.0126516
- Liu B, Berr A, Chang C, Liu C, Shen W-H, Ruan Y. Interplay of the histone methyltransferases SDG8 and SDG26 in the regulation of transcription and plant flowering and development. Biochim Biophys Acta 2016; 1859:581-90; PMID:26854085; http://dx.doi.org/10.1016/j.bbagrm.2016.02.003
- Kumar SV, Wigge PA. H2A.Z-Containing Nucleosomes Mediate the Thermosensory Response in Arabidopsis. Cell 2010; 140:136-47; PMID:20079334; http://dx.doi.org/10.1016/j.cell.2009.11.006
- Probst AV, Mittelsten Scheid O. Stress-induced structural changes in plant chromatin. Curr Opin Plant Biol 2015; 27:8-16; PMID:26042538; http://dx.doi.org/10.1016/j.pbi.2015.05.011
- Pecinka A, Dinh HQ, Baubec T, Rosa M, Lettner N, Scheid OM. Epigenetic Regulation of Repetitive Elements Is Attenuated by Prolonged Heat Stress in Arabidopsis. Plant Cell 2010; 22:3118-29; PMID:20876829; http://dx.doi.org/10.1105/tpc.110.078493
- Zemach A, Kim MY, Hsieh P-H, Coleman-Derr D, Eshed-Williams L, Thao K, Harmer SL, Zilberman D. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 2013; 153:193-205; PMID:23540698; http://dx.doi.org/10.1016/j.cell.2013.02.033
- Rutowicz K, Puzio M, Halibart-Puzio J, Lirski M, Kroteń MA, Kotliński M, Kniżewski L, Lange B, Muszewska A, Śniegowska-Świerk K, et al. A specialized histone H1 variant is required for adaptive responses to complex abiotic stress and related DNA methylation in Arabidopsis. Plant Physiol 2015; 169:2080-101; PMID:26351307; http://dx.doi.org/10.1104/pp.15.00493
- Verkest A, Byzova M, Martens C, Willems P, Verwulgen T, Slabbinck B, Rombaut D, Van de Velde J, Vandepoele K, Standaert E, et al. Selection for improved energy use efficiency and drought tolerance in canola results in distinct transcriptome and epigenome changes. Plant Physiol 2015; 168:1338-50; PMID:26082400; http://dx.doi.org/10.1104/pp.15.00155
- Costas C, de la Paz Sanchez M, Stroud H, Yu Y, Oliveros JC, Feng S, Benguria A, López-Vidriero I, Zhang X, Solano R, et al. Genome-wide mapping of Arabidopsis thaliana origins of DNA replication and their associated epigenetic marks. Nat Struct Mol Biol 2011; 18:395-400; PMID:21297636; http://dx.doi.org/10.1038/nsmb.1988
- Karnani N, Taylor CM, Malhotra A, Dutta A. Genomic study of replication initiation in human chromosomes reveals the influence of transcription regulation and chromatin structure on origin selection. Mol Biol Cell 2010; 21:393-404; PMID:19955211; http://dx.doi.org/10.1091/mbc.E09-08-0707
- Sequeira-Mendes J, Díaz-Uriarte R, Apedaile A, Huntley D, Brockdorff N, Gómez M. Transcription initiation activity sets replication origin efficiency in mammalian cells. PLoS Genet 2009; 5:e1000446; PMID:19360092; http://dx.doi.org/10.1371/journal.pgen.1000446
- Roudier F, Ahmed I, Bérard C, Sarazin A, Mary-Huard T, Cortijo S, Bouyer D, Caillieux E, Duvernois-Berthet E, Al-Shikhley L, et al. Integrative epigenomic mapping defines four main chromatin states in Arabidopsis. EMBO J 2011; 30:1928-38; PMID:21487388; http://dx.doi.org/10.1038/emboj.2011.103
- Sequeira-Mendes J, Aragüez I, Peiró R, Mendez-Giraldez R, Zhang X, Jacobsen SE, Bastolla U, Gutierrez C. The Functional Topography of the Arabidopsis Genome Is Organized in a Reduced Number of Linear Motifs of Chromatin States. Plant Cell 2014; 26:2351-66; PMID:24934173; http://dx.doi.org/10.1105/tpc.114.124578
- Swoboda P, Gal S, Hohn B, Puchta H. Intrachromosomal homologous recombination in whole plants. EMBO J 1994; 13:484; PMID:8313893; http://dx.doi.org/10.1046/j.1365-313x.1995.7020203.x
- Gao J, Zhu Y, Zhou W, Molinier J, Dong A, Shen W-H. NAP1 family histone chaperones are required for somatic homologous recombination in arabidopsis. Plant Cell 2012; 24:1437-47; PMID:22534127; http://dx.doi.org/10.1105/tpc.112.096792
- Zhang C, Cao L, Rong L, An Z, Zhou W, Ma J, Shen W-H, Zhu Y, Dong A. The chromatin-remodeling factor AtINO80 plays crucial roles in genome stability maintenance and in plant development. Plant J 2015; 82:655-68; PMID:25832737; http://dx.doi.org/10.1111/tpj.12840
- Willing E-M, Rawat V, Mandáková T, Maumus F, James GV, Nordström KJV, Becker C, Warthmann N, Chica C, Szarzynska B, et al. Genome expansion of Arabis alpina linked with retrotransposition and reduced symmetric DNA methylation. Nat Plants 2015; 1:14023; PMID:27246759; http://dx.doi.org/10.1038/nplants.2014.23
- Feng S, Cokus SJ, Schubert V, Zhai J, Pellegrini M, Jacobsen SE. Genome-wide Hi-C analyses in Wild-Type and mutants reveal High-Resolution chromatin interactions in arabidopsis. Mol Cell 2014; 55:694-707; PMID:25132175; http://dx.doi.org/10.1016/j.molcel.2014.07.008
- Wang C, Liu C, Roqueiro D, Grimm D, Schwab R, Becker C, Lanz C, Weigel D. Genome-wide analysis of local chromatin packing in Arabidopsis thaliana. Genome Res 2015; 25:246-56; PMID:25367294; http://dx.doi.org/10.1101/gr.170332.113
- Arner E, Daub CO, Vitting-Seerup K, Andersson R, Lilje B, Drabløs F, Lennartsson A, Rönnerblad M, Hrydziuszko O, Vitezic M, et al. Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science 2015; 347:1010-4; PMID:25678556; http://dx.doi.org/10.1126/science.1259418
- Chepelev I, Wei G, Wangsa D, Tang Q, Zhao K. Characterization of genome-wide enhancer-promoter interactions reveals co-expression of interacting genes and modes of higher order chromatin organization. Cell Res 2012; 22:490-503; PMID:22270183; http://dx.doi.org/10.1038/cr.2012.15
- Gross DS, Garrard WT. Nuclease Hypersensitive Sites in Chromatin. Annu Rev Biochem 1988; 57:159-97; PMID:3052270; http://dx.doi.org/10.1146/annurev.bi.57.070188.001111
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci 2010; 107:21931-6; PMID:21106759; http://dx.doi.org/10.1073/pnas.1016071107
- Zhu Y, Sun L, Chen Z, Whitaker JW, Wang T, Wang W. Predicting enhancer transcription and activity from chromatin modifications. Nucleic Acids Res 2013; 41:10032-43; PMID:24038352; http://dx.doi.org/10.1093/nar/gkt826
- Stam M, Mittelsten Scheid O. Paramutation: an encounter leaving a lasting impression. Trends Plant Sci 2005; 10:283-90; PMID:15949762; http://dx.doi.org/10.1016/j.tplants.2005.04.009
- Hövel I, Pearson NA, Stam M. Cis-acting determinants of paramutation. Semin Cell Dev Biol 2015; 44:22-32; PMID:26321497; http://dx.doi.org/10.1016/j.semcdb.2015.08.012
- Matzke MA, Kanno T, Matzke AJM. RNA-Directed DNA Methylation: The Evolution of a Complex Epigenetic Pathway in Flowering Plants. Annu Rev Plant Biol 2015; 66:243-67; PMID:25494460; http://dx.doi.org/10.1146/annurev-arplant-043014-114633
- Blevins T, Pontvianne F, Cocklin R, Podicheti R, Chandrasekhara C, Yerneni S, Braun C, Lee B, Rusch D, Mockaitis K, et al. A Two-Step Process for Epigenetic Inheritance in Arabidopsis. Mol Cell 2014; 54:30-42; PMID:24657166; http://dx.doi.org/10.1016/j.molcel.2014.02.019
- Hauben M, Haesendonckx B, Standaert E, Van Der Kelen K, Azmi A, Akpo H, Van Breusegem F, Guisez Y, Bots M, Lambert B, et al. Energy use efficiency is characterized by an epigenetic component that can be directed through artificial selection to increase yield. Proc Natl Acad Sci 2009; 106:20109-14; PMID:19897729; http://dx.doi.org/10.1073/pnas.0908755106
- De Block M, Van Lijsebettens M. Energy efficiency and energy homeostasis as genetic and epigenetic components of plant performance and crop productivity. Curr Opin Plant Biol 2011; 14:275-82; PMID:21411363; http://dx.doi.org/10.1016/j.pbi.2011.02.007
- Mirouze M, Vitte C. Transposable elements, a treasure trove to decipher epigenetic variation: insights from Arabidopsis and crop epigenomes. J Exp Bot 2014; 65:2801-12; PMID:24744427; http://dx.doi.org/10.1093/jxb/eru120
- de Paula CMP, Souza Sobrinho F, Techio VH. Chromosomal distribution of H3K4me2, H3K9me2 and 5-methylcytosine: variations associated with polyploidy and hybridization in Brachiaria (Poaceae). Plant Cell Rep 2016; 35:1-11; PMID:27015682; http://dx.doi.org/10.1007/s00299-016-1969-z