
ABSTRACT
The increase in the prevalence of allergic diseases is believed to partially depend on environmental changes. DNA methylation is a major epigenetic mechanism, which is known to respond to environmental factors. A number of studies have revealed that patterns of DNA methylation may potentially predict allergic diseases.
Here, we examined how maternal atopy is associated with methylation patterns in the cord blood of neonates.
We conducted an epigenome-wide association study in a cohort of 96 mother-child pairs. Pregnant women aged not more than 35 years old, not currently smoking or exposed to environmental tobacco smoke, who did not report obesity before conception were considered eligible. They were further tested for atopy. Converted DNA from cord blood was analysed using Infinium MethylationEPIC; for statistical analysis, RnBeads software was applied. Gestational age and sex were included as covariates in the final analysis.
83 DM sites were associated with maternal atopy. Within the top DM sites, there were CpG sites which mapped to genes SCD, ITM2C, NT5C3A and NPEPL1. Regional analysis revealed 25 tiling regions, 4 genes, 3 CpG islands and 5 gene promoters, (including PIGCP1, ADAM3A, ZSCAN12P1) associated with maternal atopy. Gene content analysis revealed pointwise enrichments in pathways related to purine-containing compound metabolism, the G1/S transition of the mitotic cell cycle, stem cell division and cellular glucose homoeostasis.
These findings suggest that maternal atopy provides a unique intrauterine environment that may constitute the first environment in which exposure is associated with methylation patterns in newborn.
Introduction
In recent decades, we have observed substantial increases in the prevalence of allergic diseases. Environmental changes and rapid development of industrialization and urbanization, together with modern lifestyle factors, are considered to be the principal factors in this process[Citation1].
The first environment to which human beings are exposed occurs in utero, where individuals are surrounded by maternal metabolic and immune milieu before birth. It has been shown that the in utero environment in asthmatic mothers is very specifically different from non-asthmatic mothers, and direct effects of the altered cytokine environment, induced by asthma, exist in pregnancy[Citation2]. Maternal effects have been described in relation to atopy and other allergy outcomes [Citation3,Citation4]. Also, epidemiological studies concerning early allergy phenotypes indicate that the prenatal period is the part of life that seems to be most crucial for promoting immune system maturation [Citation5,Citation6]. Both atopy and asthma begin in utero [Citation2,Citation7]. This observation is similar to those made in other studies, where prenatal programming conditioned disease phenotypes, such as diabetes or metabolic syndrome, later in life [Citation8,Citation9].
DNA methylation (DNAm) is a major epigenetic mechanism that influences gene expression. However, this relationship is complicated and not fully understood. DNAm is known to respond to environmental factors and connects fundamental mechanisms of genetics and the environment to allergy development, with both genes and the environment mediating interactions on the molecular level. A number of studies have revealed that the DNAm patterns of allergy genes are associated with allergic diseases [Citation10–13].
Many factors contribute to DNAm changes in the foetus. Exposure to maternal smoking[Citation14], BMI[Citation15], DHA levels[Citation16], vitamin D levels [Citation17] and folate supplementation [Citation18] have been shown to be associated with DNAm in cord blood. Thus, we hypothesized that maternal atopy influences prenatal programming.
Here we present genome-wide DNAm patterns in cord blood in a cohort of newborns in relation to the occurrence of maternal atopy to identify both general methylation patterns and the state of specific genes that are differentially methylated.
Results
All recruited pregnant women were tested for atopy at enrolment. Apart from having or not having pets, there were no significant differences between atopic and non-atopic mothers. In the atopic group, asthma was reported in 12%, allergic rhinitis in 30% and atopic dermatitis in 20%. The mean age was 29 years in both the atopic and non-atopic groups. The mean BMI was 20.5 in the atopic group and 21.5 in the non-atopic group, and the mean GWG (gestational weight gain) was 14.81 and 12.9, respectively, in the atopic and non-atopic groups. The proportion of caesarean sections was relatively high: 42% and 47%, respectively. In comparison, in the general population, the national proportion of caesarean sections in the year 2017 was 44% (https://ec.europa.eu/eurostat/). The majority of women were highly educated – they graduated from University with a degree. In both groups, more than 80% of women were taking vitamin supplements – the majority being multivitamins which contain vitamin D and folate. Characteristics of the groups studied are presented in .
Table 1. Characteristic of study group. C p < 0.05 were considered significant; A chi2 test was performed to assess categorical data, a Man-Whitney U test was used for quantitative variables. Data are presented as mean ± SD or as numbers and frequencies (%). GWG gestational weight gain
Global methylation profile differentiation
Probe filtering based on quality resulted in a final set of 827,101 interrogated methylation sites across all 96 samples. An initial test that detected an association between traits (covariates) and principal components of methylation variability within samples showed that gestational age and sex were associated with methylation profile characteristics. In the final analysis, gestational age and sex were used to correct obtained methylation β-values. Additionally, a correction for cell-type heterogeneity was implemented, as proposed by Houseman et al. (2014)[Citation19]. Differentiation of global methylation profiles of the study groups (as shown using principal component analysis) was low, with a visible subgroup of samples showing distinct methylation profiles that included subjects predominantly from non-atopic mothers (Supplementary File 1). This sub-cluster was also distinct when methylation profiles were analysed on regional levels, which corresponded to promoters, genes and Chg. islands. This sub-cluster remained separated when unsupervised hierarchical clustering was performed based on Euclidean distance (Supplementary File 2). The distribution of methylation values across different genomic regions (CpG contexts) was also similar between groups (Supplementary File 3).
Site-level differential methylation analysis
Analysis of differential methylation revealed very few significantly differentially methylated sites after FDR (p < 0.05, n = 43). Nevertheless, following the guidelines of Müller et al. [Citation20], we have used the RnBeads software score to identify DM sites with high levels of confidence. As differentially methylated, we selected the top 0.01% of sites based on the empirical distribution of their scores. This resulted in the selection of 83 sites which differed between offspring born to atopic mothers and controls with a high confidence (; ; Supplementary File 4). The analysis of site annotations showed that they were predominantly localized within gene bodies (48.2%) and intergenic regions (33.7%). Only 19.3% of the DM sites considered were located within known CpG islands. Most of the DM sites identified were hypomethylated in the atopic group (n = 49; 59%). The average absolute delta beta value for hypermethylated sites in the atopic group was low (0.054), while for hypomethylated sites it was 0.047. The distribution of hyper- and hypomethylated DM sites in different functional elements differed highly significantly (Chi-Square test p < 0.01). The hypermethylated sites were more commonly located in intergenic regions (42.4% in hyper- vs. 28.6% in hypomethylated sites), while hypomethylated sites were more common in gene bodies (55.1% in hypo- vs. 39.4% in hypermethylated sites) (Supplementary Table 1). Within the 83 significant individual sites identified, there were 25 mQTL associated CpG sites[Citation21].
Table 2. Top individual CpG sites differentially methylated between the analysed groups and their annotations in mQTL and EWAS Atlas databases
Figure 1. Plot of mean methylation difference between groups against – log10p-value for differential methylation of (a) CpG sites, (b) tailing regions, (c) genes, (d) promoters and (e) CpG islands. Colour gradient corresponds to combined rank based on RnBead software score.
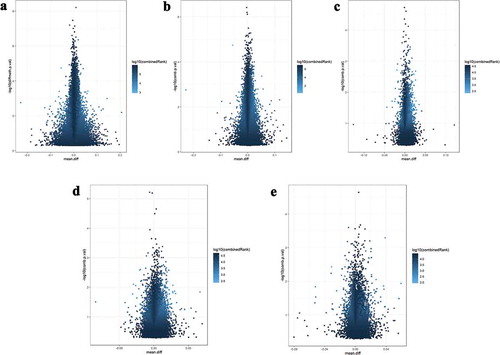
Analysis of differentially methylated regions
Region-based tests included tiling regions, genes, promoters and GpG islands. From each category, the top 0.01% of regions with the best identified RnBead scores were selected as DMRs. This resulted in the selection of 25 tiling regions, 4 genes, 3 CpG islands and 5 gene promoters, which contained at least two probes inside each region (Supplementary File 5). Most DM tiles (n = 20) were hypomethylated in the atopic group, which had an average delta beta value of 0.028. The number of sites within tiled regions ranged from 2 to 12, and the regions were distributed throughout 18 different autosomes. All gene regions that were differentially methylated were hypomethylated in the atopic group with an average delta beta value of 0.025. The genes included PIGCP1, ADAM3A, AL390719.2 and ZSCAN12P1. DM promoters with at least two analysed probes corresponded to CMYA5 and A2M-AS1 genes and AC005481.1, AC114291.1 and AC005280.1 novel transcripts. Promoters were both hypo- and hypermethylated with an average absolute delta beta value of 0.023. The three identified CpG islands had a mean absolute delta beta value of 0.032 and were localized on chromosomes 7 (158,854,447–158,854,967 bp), 9 (90,621,589–90,622,101) and 12 (9,217,329–9,217,715).
Gene content analysis of DM sites and regions
The sites that were DM between atopic and control groups were associated with 50 different genes. Together, the genes were not significantly enriched (after correction for multiple testing) in any biological processes, KEGG pathways or disease phenotypes. However, some pointwise enrichments have been observed for purine-containing compound metabolic processes (ADCY8, ESRRB, HK1, MRI1, NT5C3A, and SCD), the G1/S transition of the mitotic cell cycle (CUL3, ESRRB, NACC2, and PRIM2), stem cell division (CUL3, ESRRB) and cellular glucose homoeostasis (ADCY8, HK1, and PTPRN2). The subset of genes with hypermethylated sites (n = 16) was mainly associated with the cell cycle G1/S phase transition and DNA replication, while hypomethylated sites (n = 34) were associated with purine ribonucleotide biosynthetic processes (Supplementary File 6).
The tiling regions between atopic and control groups that were identified as DM spanned 19 different genes. These genes did not enrich any biological processes after FDR; however, pointwise significance was found for cell death regulation (NCK adaptor protein 2, EYA4, MEAK7, GRIN2A, DAPK1, GRK5), the glutamate receptor signalling pathway (GRIN2A, DAPK1) or phenotypes associated with sudden cardiac death (EYA4, DTNA) (Supplementary File 7). DM regions covering known CpG islands spanned two different genes, namely VIPR2 and LINC00612, which, like genes and promoters identified via regional tests, were either not classified in GO databases or were not enriched in any annotation categories.
Discussion
This study demonstrated that maternal atopy is associated with specific epigenetic signatures in offspring. Individual site analysis identified 83 CpG sites which map to 50 genes associated with maternal atopy. The most significantly differentially methylated sites annotated to specific genes included C20orf166, STAC, SYT8, KCNJ15, SCD, LINCOO669, PLEKHA2, ITM2C, NT5C3A and NPEPL1. The range of methylation differences were 5–22%. Within this group, DMs mapped to four genes known to impact the immune system, allergy and asthma. These identified genes were SCD, ITM2C, NT5C3A and NPEPL1.
The SCD gene encodes a stearoyl-coenzyme A desaturase, which has been reported to associate with two major allergic phenotypes: atopic dermatitis and asthma. The gene plays an important role in lipid synthesis. Disrupted expression of SCD is associated with patients presenting atopic dermatitis and causes a malfunction in the skin barrier of these patients[Citation22]. In experimental models of asthma, inhibition of the enzyme encoded by SCD leads to airway hyper-responsiveness and reduced immune defence against viruses[Citation23]. ITM2C is a target gene of GATA-3, a T-cell specific transcription factor. ITM2C deficiency has little impact on the function of polyclonal T cells, but attenuates T helper cell-dependent immune responses[Citation24], which seems to be specifically important for the future development of Th2 phenotypes such as allergy. Additionally, altered methylation of ITM2C, STAC, PRIM2 and CCD81 genes has been revealed in the lung cells of patients with asthma[Citation25]. NT5C3A is a gene encoding a protein that affects erythrocyte function. Recently, studies have shown a different regulatory function for the gene. NT5C3A expression is induced by IFN gamma and acts as an anti-inflammatory regulator via an epigenetic mechanism[Citation26]. IFN gamma is one of the most important cytokines affecting allergic outcomes by regulating the maturation of T-cells. Another gene, NPEPL1, is associated with FEV1/FVC ratio in smokers, implying a possible biological function for the gene in the occurrence of asthma. FEV1/FVC is the most sensitive indicator of bronchial obstruction in clinical practice. It is also widely used in research as a proxy of asthma severity and lung function[Citation27].
Furthermore, beyond the top most significantly differentially methylated sites, were others mapped to genes previously reported for their association with allergy including CCDC81[Citation28], LGR6 [Citation25,Citation29,Citation30], MIR166[Citation31], ESRRB [Citation13,Citation25,Citation32], HK1[Citation33], TNFRSF17[Citation34], PTPRN2 [Citation13,Citation25,Citation35–37], GRK5 [Citation38,Citation39], CAMTA1 [Citation40,Citation41], NCK2[Citation42], COLEC11[Citation43], VIPR2 [Citation25,Citation44–46], and PRIM2[Citation41]. Changed methylation patterns corresponding with allergic and respiratory phenotypes were also identified for SYT8 [Citation25,Citation30], TNN12, NXN, INP5A and ARHGAP30 [Citation13,Citation25], ADYC8 [Citation25,Citation47], NACC2 [Citation13,Citation25,Citation30,Citation48], SRPRB[Citation49], ERICH1 [Citation13,Citation25,Citation47], CUL3 and AHRR [Citation25]. AHRR was revealed in many studies as the most significant marker of smoking exposure in both neonate and adult populations [Citation50,Citation51]. As our group was not exposed to tobacco smoke during pregnancy, this is an unexpected result. Also, we did not observe a significant difference between groups in smoking habits before pregnancy. Possibly, tobacco exposure prior to pregnancy could play a role or some air pollutant has a similar effect.
While comparing our results with other EWAS studies, we found three CpG sites which had been previously revealed as associated with allergy. The first, cg04453550, mapped to TNFRSF17 (a 12% methylation difference was observed). Hypermethylation within this site has previously been shown to be associated with food allergies[Citation34]. TNFRSF17 protein plays a role in B-cell maturation, signal transduction, cell survival and proliferation[Citation52]. The second, cg01400671, is an intragenic region [Citation13] and the third, Cg13575925, mapped to LOC144571[Citation53]. For some other differentially methylated CpG sites identified, associations have been shown between methylation patterns and phenotypes such as ageing, maternal smoking, maternal diabetes and air pollution.
In our initial analysis, we excluded methylation markers which overlapped with SNPs. Further, we used an mQTL database for the assessment of genetic influence on methylation signatures in newborns. In the 10 most highly ranked CpG sites identified, cg12746557 mapped to KCNJ15 was mQTL related. CpG sites with lower rank within genes – CCDC81, MIR166, ESRRB and PTPRN2 – were also mQTL related. This type of epigenetic marker has been described as highly heritable and constant through and are indicative of the impact of genetics on DNAm[Citation21].
Via regional analysis, we identified 37 differentially methylated regions. These regions map to genes and gene promoters including PIGCP1, ADAM3A, ZSCAN12P1, CMYA5, A2M-AS1, as well as new transcripts including AC005481.1, AC114291.1, AC005280.1, PIGCP1, ADAM3A, ZSCAN12P1 and pseudogenes. Pseudogenes act as traps for transcripts and may affect gene regulation. ADAM33 and ZSCAN1, other members from the ADAMs and ZSCAN superfamilies, have been reported in GWAS and EWAS studies to being associated with allergic diseases [Citation54,Citation55]. However, according to current knowledge, ADAM3 protein function is related to fertility and cell-to-cell adhesion. Also, this particular member of the ADAM superfamily is lacking the metalloprotease activity [Citation56] which seems to be important according to the functional role of ADAM33 in asthma[Citation57]. There are studies referring to ADAM3A as being associated with specific neoplastic diseases, pointing to its regulatory role[Citation58]. ZSCAN proteins are transcription factors that may be involved in angiogenesis, cell apoptosis and differentiation, cell migration and cell invasion, cell proliferation and stem cell properties[Citation59], but the function of ZCAN12P1 is unknown. A2M-AS1, an RNA gene, has been reported to associate with chronic lung disease[Citation60].
In the enrichment analysis of 50 genes associated with DM sites, we found that genes did not significantly enrich any biological processes, KEGG pathways or disease phenotypes collectively. However, pointwise enrichment has been observed in purine-containing compound metabolic processes (ESRRB, HK1, NT5C3A, SCD genes), the G1/S transition of the mitotic cell cycle (ESRRB, PRIM2), stem cell division (ESRRB) and cellular glucose homoeostasis (HK1, PTPRN2). This is indicative of the complex character of variation observed.
To our knowledge, there are two studies concerning the maternal effect on DNAm at birth in the current literature. Both investigated the effects of the occurrence of maternal asthma. In the first study, 12 CpG regions within the 10 genes were described, that had methylation differences greater than 10%. The study was conducted in 12-month-old infants born to 25 asthmatic mothers and 12 mothers that were not asthmatic. There were atopic mothers in both groups; however, a higher proportion occurred in the asthmatic group. The authors determined the relationship between the methylation status of PM20D1 and atopy[Citation4]. A second study included a cohort of 36 children born to asthmatic mothers, some of whom were atopic. The authors compared methylomes at birth and prospectively evaluated the development of asthma later in life. Within this study, 589 DM regions were found[Citation55]. Both studies identified two differentially methylated genes that were significantly associated with the presence of maternal asthma, PIWIL1 and SMAD3. However, neither gene was identified in our study. Population differences could be the reason why we did not replicate these findings. In both mentioned papers, the population of mothers were mixed according to atopy status, so atopic mothers were included in asthma and non-asthma groups. Also, other parameters such as metabolic status, smoking and maternal age were not included in those analyses. Both PIWIL1 and SMAD3 are specifically related to asthma. PIWIL1 is overexpressed in asthmatic airway epithelia, SMAD3 functions as a regulator of TGF beta, thus impacting Treg and Th17 differentiation, and also correlates with the level of IL1 beta, a pro-inflammatory mediator in asthma.
This study had some advantages over previous reports. We selected a specific group of pregnant women, whose children could be estimated to be within a high-risk population for the development of allergy. These children were born in an industrialized city and to an atopic mother, but they also lacked risk factors such as pre-pregnancy obesity, metabolic complication of pregnancy or tobacco smoke exposure. These criteria made the group more homogeneous but also very unique, with no adequate currently available replication cohort. In VIVA, ALSPAC and R generation cohorts, the data for maternal atopy were collected as self-reported asthma, allergic rhinitis or eczema, which are slightly different phenotypes[Citation48]. We performed our analysis while considering factors previously reported to impact methylation in cord blood, such as maternal age, BMI[Citation61], GWG [Citation62] and healthy diet index[Citation63]. Also, we have applied cell heterogeneity adjustments in cord blood[Citation19], which increased the reliability of our results in comparison to unadjusted analyses. We used the RefFreeEWAS method proposed by Houseman et al. i. This method was shown to work equally well as other methods using reference datasets for cord blood[Citation64]. Additionally, according to Houseman et al., RefFreeEWAS, may adjust for detailed cell type differences that may be unavailable even in existing reference datasets [Citation65]. Umbilical cord blood contains mixed leukocyte populations with nucleated red blood cells. The latter is only a minor fraction of the cord blood cell population (3.4–9.3%) [Citation66] but could increase due to prenatal stress or hypoxia in situations such as maternal smoking, obesity or preterm birth. As our mothers were not smoking, not obese and in general had children born on term, these complications were not the case in our group. Presumably, variations in nucleated red blood cells should not significantly affect global methylation profiles.
In this study, the FDR method was used for multiple testing correction. However, we decided to use RnBeads ‘combined rank’ as it combines statistical significance as well as size and quotient of methylation difference, which presumably strengthens differential methylation analysis. In our results, 43 sites had significant differences after FDR between groups; however, they poorly covered sites identified by rank analysis. The sites which were significant after FDR had a mean methylation difference among groups of 2.5E-05. The calculation of combined rank ‘addresses the problem that minimal but consistent differences tend to achieve low p-value that doesn’t reflect biological significance.’[Citation67]
We have used allergic sensitization as a proxy for atopy. It is an objective measure and the very first allergic outcome reflecting an alternation in the immune system. Atopy, in consequence, leads to allergic symptoms, such as asthma, allergic rhinitis or atopic dermatitis. However, not all atopic individuals have clinical manifestations of inappropriate IgE immune response. In some epidemiological studies, up to 43% of subjects sensitized to common inhalant allergens did not present any respiratory symptoms[Citation68]. But, on the other hand, IgE from asymptomatic subjects have been revealed as functional, meaning that they initiate the cascade of cells, cytokine and inflammation even in the absence of symptoms [Citation69–71].
The majority of our group of pregnant women had higher education. Possibly, this group of people are more prone to participate in scientific research but, also, they are attending birth schools more eagerly than others. The proportion is high in the general population anyway; in the year 2018, 44% of young people graduated from tertiary educational institutions in our country[Citation72].
We have shown that maternal atopy is associated with DNAm in cord blood and that methylation patterns are clearly distinguishable between neonates born to atopic and non-atopic mothers. Although the observed methylation differences in the majority of sites are not very high (2–22%), we considered that a cumulative effect of methylation was likely to occur.
Methods
Study population
Study participants were recruited as a part of the ongoing ELMA – Epigenetic Hallmark of Maternal Atopy and Diet study, a prospective study of women and their children. ELMA was established to identify exposures that may contribute to allergic disease susceptibility in children. Women living in the metropolitan area, not currently smoking or exposed to ETS (environmental tobacco smoke), who did not report obesity before conception, gestational diabetes or hypertension were considered eligible and were recruited during their third trimester (≥28 week of gestational age) of pregnancy by attendants of a childbirth schools situated in obstetrics clinics and hospitals in Wroclaw. From November 2016 to January 2019, 158 women were enrolled and the majority (72.15%) were successfully followed to delivery. Umbilical cord blood samples were collected by midwifes or obstetricians at birth. Children from these pregnancies are being followed at 3, 6 and 12–18 months of age.
Allergic sensitization (atopy) in mothers was defined as the presence of sensitization to allergens via measurement of serum allergen-specific immunoglobulin E (IgE) for 20 allergens including cow milk, egg, wheat flour, peanuts, dust mites, trees, grasses, cat, dog, Alternaria and Cladosporium allergens (POLYCHECK, Biocheck, Germany). Women were designated atopic if they displayed at least one positive reaction (sIGE ≥0.35kUL−1) to any allergen.
All women completed food frequency questionnaires FFQ containing queries about diet that was based on the FFQ KOMPAN questionnaire. The questionnaire included 38 questions regarding the estimated frequency of intake of food groups and beverages. Finally, analyses were performed using a derivative variable (healthy diet index), which was based on the frequency of whole meal bread, buckwheat, oatmeal, whole grain pasta or other coarse cereal, milk, fermented milk drink (yoghurt, kefir, cottage cheese), white meat (chicken, turkey, rabbit, fish), legume seed (bean, pea, soybean, lentil), fruit and vegetable consumption[Citation73]. The index is ranged from 0 to 20, with higher values reflecting a healthier diet.
The cord blood samples from 96 mother-child pairs were selected for DNAm analysis, 50 from atopic mothers and 46 from mothers that were non-atopic. The selection of probes was based on DNA availability and quality. Mothers aged more than 35 years were excluded from this analysis.
The study was approved by the Ethical Committee of the Wroclaw Medical University and all participants signed informed consent forms.
Methylome analysis
Cord whole blood was secured in EDTA probes during delivery, according to standard procedures. DNA was extracted from whole blood using the QIAamp DNA Blood Mini Kit (Qiagen), according to the manufacturer’s protocol. DNA was further quantified using a Qubit 2.0 fluorimeter. DNA probes with unsatisfying quality were excluded. DNA was converted for bisulphite sequencing using the EZ DNA Methylation™ Kit (Zymo Research). Converted DNA from 96 cord blood samples from newborns with atopic (n = 50) and non-atopic control (n = 46) mothers were finally analysed using the Infinium MethylationEPIC Kit (Illumina, San Diego, CA). The hybridized and stained EPIC arrays were ultimately scanned using a HiScanSQ system (Illumina).
Infinium MethylationEPIC arrays covered over 850,000 methylation sites across the human genome. Apart from sites within known CpG islands, they included probes identifying CpG sites outside CpG islands, non-CpG methylated sites identified in human stem cells (CHH sites), DMs identified in tumour versus normal cells, FANTOM5 enhancers, ENCODE open chromatin and enhancers, DNase hypersensitive sites and sites located in miRNA promoter regions.
Data analysis
Intensities obtained after probe scanning were added to strictly control quality, filtering and normalization. Initial quality control was performed via the analysis of array controls using the BeadArray Reporter Software (Illumina). These analyses did not reveal any significant deviations from expected values. Further filtering was accomplished using RnBeads software[Citation74]. Initially, Infinium probes overlapped with SNPs, and sites with missing β values were removed. Both sites and samples were filtered using a Greedy approach, which iteratively removes probes and samples of highest impurity from the dataset. These correspond to the rows in the detection p-value table that contain the largest fraction of unreliable measurements. Additionally, probes located on sex chromosomes were excluded as a result of expected variation resulting from sex differences. The obtained data were normalized using the BMIQ procedure [Citation75] and batch effects, such as array and array position as well as other hidden confounders, were identified and removed using the surrogate variable analysis (SVA) method[Citation76]. The significance of covariates including healthy diet index, age, BMI, gestational age, birth weight, GWG, parity, type of delivery, having pets and sex were evaluated using a procedure that was also implemented in RnBeads software. While only gestational age and sex traits were significantly associated with methylation of samples, they were included in a final analysis in which the refFreeEWAS method [Citation65] was used for beta-value (methylation values) corrections regarding cell-type heterogeneity and the inclusion of the two mentioned covariates. The refFreeEWAS method allows reference-free deconvolution that provides proportions of putative cell types defined by their underlying methylomes and allows an explicit quantitation of the mediation of phenotypic associations with DNAm by cell composition effects. Finally, RnBead software scores were used to detect sites that were differentially methylated between the groups considered. RnBeads combines statistical testing with a priority ranking scheme that is based on the absolute and relative effect size of the differences between groups, and it assigns a combined rank score for differential DNAm to each analysed CpG site and genomic region. This combined rank is defined as the maximum (i.e., worst) of three individual rankings: (i) by absolute difference in mean DNAm levels, (ii) by the relative difference in mean DNAm levels, which is calculated as the absolute value of the logarithm of the quotient of mean DNAm levels, and (iii) by the CpG-based or region-based p-value, calculated as described above[Citation67].
The smaller the combined rank for a site, the greater the evidence for differential methylation exhibited. The top 0.01% of differentially methylated sites with best ranks were analysed as well as the top 0.01% of differentially methylated regions (DMRs). DMR types were defined as tiling regions (with a window size of 5 kb across the genome), genes, promoters (Ensembl gene definitions were used that define promoter regions as the 1,500 bases upstream and 500 bases downstream of the transcription start sites of corresponding genes) and GpG islands (obtained from tracks in UCSC Genome Browser). The genes associated with DM sites and DM regions were analysed using the WEB-based GEne SeT AnaLysis Toolkit [Citation77] to identify enriched biological processes, pathways and disease phenotypes. Overrepresentation tests were performed with respect to all known genes (genome) using an FDR [Citation78] correction for multiple testing. Additional gene annotations were performed using the Panther Classification System[Citation79].
Additionally, we have performed a search using the EWAS ATLAS database [Citation80] to identify previous epigenetic studies investigating atopy- and respiratory-related phenotypes.
Conclusion
In this study, we have identified several DM CpG sites and regions in cord blood of infants born to atopic mothers. These DM sites map to genes previously revealed to be associated with allergic phenotypes and also to new ones, which can act as a focus of new studies. These findings suggest that maternal atopy constitutes a unique intrauterine environment that is associated with methylation patterns.
Supplemental Material
Download Zip (941.5 KB)Disclosure statement
Danielewicz H, Gurgul A, Dębińska A, Myszczyszyn G, Szamtoła T, Myszkal A, Jasielczuk G, Drabik-Chmaerska A report grant and personal fees from the National Science Center, Poland, during the conduct of the study; Danielewicz H reports lecturer fees from Mead Johnson Nutrition outside the submitted work; Hirnle L, Boznanski A report a grant from the National Science Center, Poland, during the conduct of the study.
Supplementary material
Supplemental data for this article can be accessed here.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Haahtela T. A biodiversity hypothesis. Allergy [Internet]. 2019 [cited 2020 Mar 17];74:1445–1456. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30835837
- Rothers J, Stern DA, Lohman IC, et al. Maternal cytokine profiles during pregnancy predict asthma in children of mothers without asthma. Am J Respir Cell Mol Biol [Internet]. 2018 [cited 2018 Dec 28];59:592–600. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29863910
- Moffatt C. Maternal effects in atopic disease. Clin Exp Allergy [Internet]. 1998 [cited 2019 Oct 4];28:56–61.
- Gunawardhana LP, Baines KJ, Mattes J, et al. Differential DNA methylation profiles of infants exposed to maternal asthma during pregnancy. Pediatr Pulmonol. 2014;49:852–862.
- Karvonen A, Lampi J, Keski-Nisula L, et al. Farm environment during pregnancy and childhood and polysensitization at the age of 31 – prospective birth cohort study in Finland. J Investig Allergol Clin Immunol. 2019;31. DOI:10.18176/jiaci.0455
- Depner M, Ege MJ, Genuneit J, et al. Atopic sensitization in the first year of life. J Allergy Clin Immunol [Internet]. 2013 [cited 2017 Mar 16];131:781–788.e9. Available from: http://www.jacionline.org/article/S0091-6749(12)01975-6/pdf
- Grieger JA, Clifton VL, Tuck AR, et al. In utero programming of allergic susceptibility. Int Arch Allergy Immunol. 2016;169:80–92.
- Danielewicz H, Myszczyszyn G, Dębińska A, et al. Diet in pregnancy—more than food. Eur J Pediatr. 2017;176: 1573-1579.
- Saad MI, Abdelkhalek TM, Haiba MM, et al. Maternal obesity and malnourishment exacerbate perinatal oxidative stress resulting in diabetogenic programming in F1 offspring. J Endocrinol Invest [Internet]. 2016 [cited 2017 Mar 11];39:643–655. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26667119
- DeVries A, Vercelli D. Epigenetics in allergic diseases. Curr Opin Pediatr [Internet]. 2015 [cited 2019 Oct 4];27:719–723. Available from: http://content.wkhealth.com/linkback/openurl?sid=WKPTLP:landingpage&an=00008480-201512000-00012
- Everson TM, Lyons G, Zhang H, et al. DNA methylation loci associated with atopy and high serum IgE: A genome-wide application of recursive random forest feature selection. Genome Med. 2015;7: 89.
- Chen W, Wang T, Pino-Yanes M, et al. An epigenome-wide association study of total serum IgE in hispanic children. J Allergy Clin Immunol. 2017;140:571–577.
- Forno E, Wang T, Qi C, et al. DNA methylation in nasal epithelium, atopy, and atopic asthma in children: a genome-wide study. Lancet Respir Med [Internet]. 2019 [cited 2019 Oct 23];7:336–346. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30584054
- Ivorra C, Fraga MF, Bayón GF, et al. DNA methylation patterns in newborns exposed to tobacco in utero. J Transl Med. 2015;13:25.
- Hjort L, Martino D, Grunnet LG, et al. Gestational diabetes and maternal obesity are associated with epigenome-wide methylation changes in children. JCI Insight. 2018;3: e122572.
- Dilli D, Doğan NN, MŞ İ, et al. MaFOS-GDM trial: maternal fish oil supplementation in women with gestational diabetes and cord blood DNA methylation at insulin like growth factor-1 (IGF-1) gene. Clin Nutr ESPEN. 2018;23:73–78.
- Cho HJ, Sheen YH, Kang MJ, et al. Prenatal 25-hydroxyvitamin D deficiency affects development of atopic dermatitis via DNA methylation. J Allergy Clin Immunol. 2019;143:1215–1218.
- Amarasekera M, Martino D, Ashley S, et al. Genome-wide DNA methylation profiling identifies a folate-sensitive region of differential methylation upstream of ZFP57-imprinting regulator in humans. Faseb J. 2014;28:4068–4076.
- Houseman EA, Molitor J, Marsit CJ. Reference-free cell mixture adjustments in analysis of DNA methylation data. Bioinformatics [Internet]. 2014 [cited 2019 Jul 22];30:1431–1439. Available from: https://academic.oup.com/bioinformatics/article-lookup/doi/10.1093/bioinformatics/btu029
- Müller F, Scherer M, Assenov Y, et al. RnBeads 2.0: comprehensive analysis of DNA methylation data. Genome Biol [Internet]. 2019 [cited 2020 Jun 26];20:55. Available from:: https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1664-9
- Gaunt TR, Shihab HA, Hemani G, et al. Systematic identification of genetic influences on methylation across the human life course. Genome Biol. 2016;17:61.
- Danso M, Boiten W, van Drongelen V, et al. Altered expression of epidermal lipid bio-synthesis enzymes in atopic dermatitis skin is accompanied by changes in stratum corneum lipid composition. J Dermatol Sci [Internet]. 2017 [cited 2019 Sep 30];88:57–66. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28571749
- Rodriguez-Perez N, Schiavi E, Frei R, et al. Altered fatty acid metabolism and reduced stearoyl-coenzyme a desaturase activity in asthma. Allergy. 2017 [cited 2019 Sep 30];72:1744–1752. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28397284
- Tai T-S, Pai S-Y, Ho I-C. Itm2a, a target gene of GATA-3, plays a minimal role in regulating the development and function of T cells. PLoS One. 2014 [cited 2019 Sep 30];9:e96535.
- Nicodemus-Johnson J, Myers RA, Sakabe NJ, et al. DNA methylation in lung cells is associated with asthma endotypes and genetic risk. JCI Insight [Internet]. 2016 [cited 2019 Oct 23];1:e90151. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27942592
- Al-Haj L, Khabar KSAA. The intracellular pyrimidine 5′-nucleotidase NT5C3A is a negative epigenetic factor in interferon and cytokine signaling. Sci Signal [Internet]. 2018 [cited 2019 Sep 30];11:eaal2434. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29463777
- Lutz SM, Cho MH, Young K, et al. A genome-wide association study identifies risk loci for spirometric measures among smokers of European and African ancestry. BMC Genet [Internet]. 2015 [cited 2019 Nov 9];16:138. Available from: http://www.biomedcentral.com/1471-2156/16/138
- Rothenberg ME, Spergel JM, Sherrill JD, et al. Common variants at 5q22 associate with pediatric eosinophilic esophagitis. Nat Genet [Internet]. 2010 [cited 2019 Nov 9];42:289–291. Available from: http://www.nature.com/articles/ng.547
- Kim S, Back SK, Na HS, et al. Capsaicin induces atopic dermatitis-like manifestations through dysregulation of proteolytic system and alteration of filaggrin processing in rats. Exp Dermatol [Internet]. 2018 [cited 2019 Sep 30];27:332–339.
- Cardenas A, Sordillo JE, Rifas-Shiman SL, et al. The nasal methylome as a biomarker of asthma and airway inflammation in children. Nat Commun [Internet]. 2019 [cited 2019 Oct 23];10:3095. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31300640
- Tian W, Du Y, Ma Y, et al. miR663a‑TTC22V1 axis inhibits colon cancer metastasis. Oncol Rep [Internet]. 2019 [cited 2019 Oct 25];41:1718–1728. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30664167
- Naranbhai V, Fletcher HA, Tanner R, et al. Distinct transcriptional and anti-mycobacterial profiles of peripheral blood monocytes dependent on the ratio of monocytes: lymphocytes. EBioMedicine [Internet]. 2015 [cited 2019 Sep 30];2:1619–1626. Available from: https://linkinghub.elsevier.com/retrieve/pii/S2352396415301444
- Everson TM, Zhang H, Lockett GA, et al. Epigenome-wide association study of asthma and wheeze characterizes loci within HK1. Allergy, Asthma Clin Immunol [Internet]. 2019 [cited 2019 Sep 30];15:43. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31367216
- Martino D, Joo JE, Sexton-Oates A, et al. Epigenome-wide association study reveals longitudinally stable DNA methylation differences in CD4+ T cells from children with IgE-mediated food allergy. Epigenetics [Internet]. 2014 [cited 2019 Oct 23];9:998–1006. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24762976
- den Dekker HT, Burrows K, Felix JF, et al. Newborn DNA-methylation, childhood lung function, and the risks of asthma and COPD across the life course. Eur Respir J [Internet]. 2019 [cited 2019 Sep 30];53:1801795. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30765504
- Quraishi BM, Zhang H, Everson TM, et al. Identifying CpG sites associated with eczema via random forest screening of epigenome-scale DNA methylation. Clin Epigenetics [Internet]. 2015;7:68. Available from: http://www.clinicalepigeneticsjournal.com/content/7/1/68%5Cnhttp://www.ncbi.nlm.nih.gov/pubmed/26199674%5Cnhttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC4508804
- North ML, Jones MJ, MacIsaac JL, et al. Blood and nasal epigenetics correlate with allergic rhinitis symptom development in the environmental exposure unit. Allergy Eur J Allergy Clin Immunol [Internet]. 2018 [cited 2019 Oct 21];73:196–205.
- Serafin DS, Allyn B, Sassano MF, et al. Chemerin-activated functions of CMKLR1 are regulated by G protein-coupled receptor kinase 6 (GRK6) and β-arrestin 2 in inflammatory macrophages. Mol Immunol [Internet]. 2019 [cited 2019 Sep 30];106:12–21. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30576947
- Dellon ES, Selitsky SR, Genta RM, et al. Gene expression-phenotype associations in adults with eosinophilic esophagitis. Dig Liver Dis [Internet]. 2018 [cited 2019 Nov 10];50:804–811. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29628359
- Baron U, Floess S, Wieczorek G, et al. DNA demethylation in the humanFOXP3 locus discriminates regulatory T cells from activated FOXP3+ conventional T cells. Eur J Immunol [Internet]. 2007 [cited 2019 Sep 30];37:2378–2389.
- Martino D, Dang T, Sexton-Oates A, et al. Blood DNA methylation biomarkers predict clinical reactivity in food-sensitized infants. J Allergy Clin Immunol [Internet]. 2015;135:1319–1328e12.
- Paensuwan P, Ngoenkam J, Khamsri B, et al. Evidence for inducible recruitment of Wiskott-Aldrich syndrome protein to T cell receptor-CD3 complex in Jurkat T cells. Asian Pacific J Allergy Immunol [Internet]. 2015 [cited 2019 Sep 30];33:189–195. Available from: http://apjai.digitaljournals.org/index.php/apjai/article/download/1878/1219
- Chang J-C, Kuo H-C, Hsu T-Y, et al. Different genetic associations of the IgE production among fetus, infancy and childhood. PLoS One. 2013 [cited 2019 Sep 30];8:e70362. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23936416
- Wang Y, Mei Y, Bao S, et al. Vasoactive intestinal polypeptide enhances oral tolerance by regulating both cellular and humoral immune responses. Clin Exp Immunol [Internet]. 2007 [cited 2019 Sep 30];148:178–187. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17349016
- Kim D-H, Park I-H, Cho J-S, et al. Alterations of vasoactive intestinal polypeptide receptors in allergic rhinitis. Am J Rhinol Allergy [Internet]. 2011 [cited 2019 Sep 30];25:e44–7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21711977
- Sundar IK, Yin Q, Baier BS, et al. DNA methylation profiling in peripheral lung tissues of smokers and patients with COPD. Clin Epigenetics [Internet]. 2017 [cited 2019 Nov 9];9:38. Available from:: http://www.ncbi.nlm.nih.gov/pubmed/28416970
- Langie SAS, Szarc Vel Szic K, Declerck K, et al. Whole-genome saliva and blood DNA methylation profiling in individuals with a respiratory allergy. PLoS One. 2016 [cited 2019 Oct 23];11:e0151109. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26999364
- Peng C, Van Meel ER, Cardenas A, et al. Epigenome-wide association study reveals methylation pathways associated with childhood allergic sensitization. Epigenetics [Internet]. 2019 [cited 2019 Oct 21];14:445–466. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30876376
- Lee MK, Hong Y, Kim S-Y, et al. Epigenome-wide association study of chronic obstructive pulmonary disease and lung function in Koreans. Epigenomics [Internet]. 2017 [cited 2019 Nov 6];9:971–984. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28621160
- Devries A, Vercelli D. The neonatal methylome as a gatekeeper in the trajectory to childhood asthma. Epigenomics. 2017;9:585–593.
- Imboden M, Wielscher M, Rezwan FI, et al. Epigenome-wide association study of lung function level and its change. Eur Respir J [Internet]. 2019 [cited 2019 Sep 30];54:1900457. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31073081
- Coquery CM, Erickson LD. Regulatory roles of the tumor necrosis factor receptor BCMA. Crit Rev Immunol [Internet]. 2012 [cited 2020 Mar 21];32:287–305. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23237506
- Langie SAS, Szic KS, Declerck K, et al. Whole-Genome saliva and blood DNA methylation profiling in individuals with a respiratory allergy. PLoS One. 2016;11. DOI:10.1371/journal.pone.0151109
- Pino-Yanes M, Corrales A, Cumplido J, et al. Assessing the validity of asthma associations for eight candidate genes and age at diagnosis effects. PLoS One. 2013;8: e73157.
- Devries A, Wlasiuk G, Miller SJ, et al. Epigenome-wide analysis links SMAD3 methylation at birth to asthma in children of asthmatic mothers. J Allergy Clin Immunol [Internet]. 2016 [cited 2018 May 4]. Available from: https://spiral.imperial.ac.uk:8443/bitstream/10044/1/43732/2/1-s2.0-S0091674916324599-main.pdf
- Primakoff P, Myles DG. The ADAM gene family: surface proteins with adhesion and protease activity. Trends Genet. 2000;16:83–87.
- Holgate ST, Davies DE, Rorke S, et al. ADAM 33 and its association with airway remodeling and hyperresponsiveness in asthma. Clin Rev Allergy Immunol. 2004;27:23–34.
- Vizcaino MA, Tabbarah AZ, Asnaghi L, et al. ADAM3A copy number gains occur in a subset of conjunctival squamous cell carcinoma and its high grade precursors. Hum Pathol [Internet]. 2019;94:92–97.
- Huang M, Chen Y, Han D, et al. Role of the zinc finger and SCAN domain-containing transcription factors in cancer. Am J Cancer Res. 2019;9:816–836.
- Qian Y, Mao Z, Shi Y, et al. Comprehensive analysis of miRNA-mRNA-lncRNA networks in non-smoking and smoking patients with chronic obstructive pulmonary disease. Cell Physiol Biochem [Internet]. 2018 [cited 2019 Oct 5];50:1140–1153. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30355907
- Haertle L, El Hajj N, Dittrich M, et al. Epigenetic signatures of gestational diabetes mellitus on cord blood methylation. Clin Epigenetics [Internet]. 2017 [cited 2018 Apr 28];9:28. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28360945
- Hrolfsdottir L, Schalkwijk CG, Birgisdottir BE, et al. Maternal diet, gestational weight gain, and inflammatory markers during pregnancy. Obesity [Internet]. 2016 [cited 2017 Sep 5];24:2133–2139. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27581164
- Lee H, Hernandez-Vargas H, Biessy C, et al. Modulation of epigenetic states and infant immune system by dietary supplementation with (Omega)-3 polyunsaturated fatty acid during pregnancy in an intervention study. Eur J Cancer [Internet]. 2012;48:S137–8. Available from: http://ovidsp.ovid.com/ovidweb.cgi?T=JS&PAGE=reference&D=emed10&NEWS=N&AN=70820354
- Kaushal A, Zhang H, Karmaus WJJ, et al. Comparison of different cell type correction methods for genome-scale epigenetics studies. BMC Bioinformatics. 2017;18:1–12.
- Houseman EA, Kile ML, Christiani DC, et al. Reference-free deconvolution of DNA methylation data and mediation by cell composition effects. BMC Bioinformatics [Internet]. 2016 [cited 2019 Jul 22];17:259. Available from: http://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-016-1140-4
- McCarthy JM, Capullari T, Thompson Z, et al. Umbilical cord nucleated red blood cell counts: normal values and the effect of labor. J Perinatol. 2006;26:89–92.
- Assenov Y, Müller F, Lutsik P, et al. Comprehensive analysis of DNA methylation data with RnBeads. Nat Methods [Internet]. 2014 [cited 2020 Apr 1];11:1138–1140. Available from: http://www.nature.com/doifinder/10.1038/nmeth.3115%5Cnpapers3://publication/doi/10.1038/nmeth.3115
- Kerkhof M, Schouten JP, De Monchy JGR. The association of sensitization to inhalant allergens with allergy symptoms: the influence of bronchial hyperresponsiveness and blood eosinophil count. Clin Exp Allergy. 2000;30:1387–1394.
- Bousquet J, Anto JM, Bachert C, et al. Factors responsible for differences between asymptomatic subjects and patients presenting an IgE sensitization to allergens. A GA2LEN project. Allergy [Internet]. 2006 [cited 2020 Mar 24];61:671–680.
- Zidarn M, Robič M, Krivec A, et al. Clinical and immunological differences between asymptomatic HDM-sensitized and HDM-allergic rhinitis patients. Clin Exp Allergy. 2019;49:808–818.
- Ciprandi G, Buscaglia S, Pesce G, et al. Minimal persistent inflammation is present at mucosal level in patients with asymptomatic rhinitis and mite allergy. J Allergy Clin Immunol [Internet]. 1995 [cited 2020 Jan 25];96:971–979. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0091674995702350
- OECD(2019). Education at a glance 2019. OECD indicators 2019. Paris: OECD Publishing; p. 1–8.
- Jeżewska- Zychowicz M, Gawęcki J, Wądołowska L, et al. Kwestionariusz do badania poglądów i zwyczajów żywieniowych dla osób w wieku od 16 do 65 lat, wersja 1.2 –kwestionariusz do samodzielnego wypełnienia przez respondenta. Rozdz. 2. Wyd Kom Nauk O Żywieniu Człowieka Pol Akad Nauk [Internet]. 2018:21–33. Available from: http://www.knozc.pan.pl/
- Müller F, Scherer M, Assenov Y, et al. RnBeads 2.0: comprehensive analysis of DNA methylation data. Genome Biol. 2019;20:55.
- Teschendorff AE, Marabita F, Lechner M, et al. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 2013;29:189–196.
- Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLOS Genet. 2007;3:1–12.
- Wang J, Vasaikar S, Shi Z, et al. WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017;45:W130–7.
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995;57:289–300.
- Mi H, Muruganujan A, Huang X, et al. Protocol update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat Protoc. 2019;14:703–721.
- Li M, Zou D, Li Z, et al. EWAS Atlas: a curated knowledgebase of epigenome-wide association studies. Nucleic Acids Res [Internet]. 2019 [cited 2019 Oct 25];47:D983–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30364969