
ABSTRACT
Despite more than 2 million American cocaine users monthly, there is no approved drug for treating cocaine use disorder. Cocaine use disorder has a multifactorial aetiology, including both genetic and environmental factors. Both cocaine use and genetic variations demonstrably alter DNA methylation and gene expression in the brain in a complex manner. How these factors interact in the context of cocaine abuse in humans is unknown. We propose that we can identify potential drug targets for treating cocaine use disorders by examining genetic, epigenetic, and expression changes in the brains of individuals that abused cocaine. In this study, we identified the interaction between the epigenetics changes (DNA CpG methylation) and genetic variants (SNPs) in the HTR2A gene in the context of cocaine addiction by using brain tissue collected from individuals that overdosed on cocaine (N = 14) and healthy matched controls (N = 16). We generated DNA CpG methylation profiles in eight regions of HTR2A harbouring frequent SNPs, measuring both allelic and total methylation, and compared these methylation profiles with HTR2A mRNA expression. Furthermore, we examined the influence of common variants rs6311 and rs6313 on cocaine abuse, methylation, and gene expression. We found evidence that rs6311 regulates HTR2A methylation, consistent with earlier studies. Furthermore, the minor alleles for rs6311 and rs6313 are associated with significantly increased expression of a splice isoform in which exon 2 is truncated in both cocaine and control samples. These results reveal specific roles for HTR2A in the context of cocaine abuse, highlighting opportunities to modulate this target for treating cocaine use disorder.
Introduction
Addiction disorders comprise a substantial economic burden in the United States, totalling more than $740 billion annually for economic and societal costs [Citation1]. Substance use disorder, as addiction is recognized clinically, is a global epidemic with a high rate of morbidity and increasing mortality rates for cocaine. Despite heavy economic and societal costs, there is no FDA-approved pharmacological treatment for cocaine addiction [Citation2]. Identifying druggable targets for new anti-addiction treatments is a major focus for addressing this significant unmet medical need.
Cocaine is a psychostimulant that blocks reuptake transporters for dopamine (DA), serotonin (5-HT), and norepinephrine (NE) neurotransmitters, which decrease their clearance from the neuronal synapse, allowing them to signal longer at pre- and post-synaptic neurotransmitter receptors [Citation3,Citation4]. Cocaine’s interference in the reuptake of 5-HT influences behavioural aspects of cocaine addiction, such as reinforcement and sensitization, making the 5-HT system a possible target for treating cocaine addiction [Citation5]. Here, we propose to address the lack of approved drugs for cocaine addiction by assessing the role of 5-HT2A receptor genetics in the context of cocaine addiction to determine its utility as an anti-addiction drug target. This receptor is readily druggable, as demonstrated by the crucial role it plays in treating psychiatric disorders [Citation6], where the selective 5-HT2A inverse agonist pimavanserin is approved by the FDA for the treatment of symptoms of delusion and hallucinations of Parkinson’s disease psychosis (PDP) [Citation7]. Preclinical studies have also demonstrated that antagonism of this receptor can attenuate addiction behaviours in rodents [Citation8].
5-HT2A is a G-protein coupled receptor most heavily expressed throughout the cerebral cortex on presynaptic and postsynaptic neuron terminals. This includes brain regions that are involved in the reward system, implicating it in drug dependence. The 5-HT2A receptor is a drug target for antipsychotic, antidepressant, and anti-anxiety drugs [Citation9]. This receptor is encoded by HTR2A, located on chromosome 13q14.2. As it is currently annotated, HTR2A consists of four exons, spanning approximately 65 kilobases. Exon 2 is alternatively spliced in human and rodent brain tissues, resulting in multiple RNA and protein isoforms [Citation10,Citation11]. This gene harbours multiple common single nucleotide polymorphisms (SNPs) associated with a wide range of medical, psychiatric, and cognitive phenotypes. For example, SNPs in HTR2A are associated with susceptibility to obsessive-compulsive disorder (OCD) and schizophrenia [Citation12]. Amongst the most well-studied SNPs in HTR2A are rs6311 (also known as −1438 A/G) and rs6313 (also known as −102 T/C). These SNPs most commonly associate with phenotypes treated by 5-HT2A modulating drugs, including stress, suicidal behaviour, schizophrenia, and drug addiction.
As it relates to addiction, several studies demonstrate an association between drug dependence and SNPs in HTR2A or the expression of 5-HT2A. One study that examined the contribution of 37 genes related to DA, 5-HT, and neurotrophic factor signalling in cocaine dependence, found a significant association between HTR2A and cocaine dependence, even when accounting for psychotic symptoms [Citation13]. Preclinical rodent studies showed that 5-HT2A antagonists attenuate cocaine-seeking behaviour and cocaine-evoked behaviour [Citation14–18]. Given the genetic, genomic, and behavioural associations between 5-HT2A and cocaine addiction, we propose that this readily druggable receptor is a suitable target for treating cocaine addiction. However, we lack an understanding of how functional SNPs in HTR2A contribute to addiction risk, particularly as it relates to cocaine.
Relevant to 5-HT2A signalling, epigenetics play a role in modulating the expression of HTR2A, and these epigenetic changes are associated with several disease-related phenotypes. For example, methylation at CpG sites – 1420 and – 1224 upstream of the HTR2A transcriptional start site in placental tissue is associated with a high level of attention in infants [Citation17]. Another study found HTR2A is hypomethylated at the rs6313 polymorphic site in schizophrenia and bipolar disorder [Citation18]. Early life stress, which increases the likelihood of drug use and abuse later in life, is also associated with changes in methylation at the HTR2A gene locus [Citation19].
Many studies show that cocaine robustly alters DNA methylation as a global effect, but few studies discuss the changes that occur at the level of individual genes. Understanding the effect of cocaine on methylation at the level of each gene is critical for developing a therapeutic hypothesis around a specific anti-addiction drug target. These findings, along with the aforementioned studies, show the complexity of the interaction between DNA methylation and cocaine addiction. Methylation modifications differ depending on the drug of abuse, mode of treatment, tissue under study, and gene locus. Given these complexities, it is important to study DNA methylation in the context of the disease you are attempting to understand.
The objective of our study is to identify the interaction between epigenetic changes, genetic variation, and gene expression at the HTR2A locus in the context of cocaine addiction, by using brain tissue collected from individuals that overdosed on cocaine and matched controls. We hypothesize that genetic factors and cocaine use alter CpG methylation in HTR2A, providing an opportunity to identify regulatory factors affecting HTR2A expression associated with cocaine abuse. We expect this study to provide us with additional validation of HTR2A as a potential drug target for treating cocaine addiction and could reveal biomarkers for drug abuse and 5-HT2A signalling.
Materials and methods
Sample selection and demographics
We measured DNA methylation in 30 human dorsolateral prefrontal cortex (DLPFC) samples dissected post-mortem by experienced neuropathologists at the University of Miami (FL). The 30 samples consisted of 14 individuals that overdosed on cocaine, and 16 controls matched for age, sex, race, and smoking status. We focused on the DLPFC because HTR2A is robustly expressed in this region, and we previously observed changes in HTR2A expression and methylation in this region that were correlated with common SNPs [Citation11]. For DNA methylation analysis, we selected HTR2A genomic regions encompassing common SNPs in the promoter and exon regions, where we anticipate that SNPs have a regulatory role in the expression of the gene and DNA methylation. We list these common SNPs in Table S1. Incorporating common SNPs also permits the analysis of allelic methylation, as described below. We excluded one sample from our final analyses due to inadequate sequencing depth.
Sample preparation
DNA extraction & bisulphite treatment
We extracted DNA using an organic extraction method. First, small pieces of DLPFC tissue were digested in a water bath overnight at 55°C in nuclei lysis buffer, SDS, and Proteinase K. Following digestion, we added 125 ml of saturated NaCl for each sample, followed by brief vortexing, to enhance precipitation of DNA. The samples were chilled at 4°C for one hour and then centrifuged for 5 minutes at 13,000rpm to pellet unwanted cellular debris. We transferred the supernatant containing the genomic DNA, to a new tube and added 1 ml of 95% ethanol, and inverted gently. We washed the precipitated DNA twice with cold 70% ethanol. We then resuspended the DNA in 50 μl of TE buffer. We performed an additional phenol-chloroform extraction to improve DNA purity. We bisulphite-treated 200–300 ng of genomic DNA for each sample with EZ DNA Methylation kit (Zymo, Irvine, California), according to manufacturer instructions. We eluted samples into a small volume (10 ul) to maintain a high DNA concentration for our sequencing applications.
SNP selection & PCR amplification
We studied the epigenetic signature of the HTR2A receptor gene using 8 SNPs (rs6310, rs6311, rs6312, rs6313, rs6314, rs3125, rs655854, rs731245; Table S1) located in the protein-coding regions (exons) and promoter region. Each SNP has a population average heterozygosity of more than 10% in the races we studied here. We designed our primers around exons and regulatory regions, presupposing that methylation in and around these regions would play a more evident role in regulating the expression of HTR2A. For our allele-specific methylation experiment, we chose to study rs6313 because it is significantly associated with many psychiatric diseases and drug abuse phenotypes, and is located in exon 2, where we previously observed alternative splicing and significant changes in CpG methylation correlated with genotype [Citation11]. Furthermore, we assessed the genotypic effects of 17 common variants in the HTR2A locus on methylation levels measured across amplicons noted in Table S1.
Bisulphite sequencing primers design & PCR amplification
For primer design, we converted the HTR2A reference sequence to a bisulphite-converted form using the BiSearch website (http://bisearch.enzim.hu/). We then designed eight strand-specific primer pairs around the eight SNPs mentioned above using PrimerSelect 15 program(Table S1). These eight amplicons assessed the methylation status at 51 CpGs. To avoid ambiguity between CpG methylation and the presence of an SNP that is part of a CpG dinucleotide sequence, we designed for non-CpG SNPs using the forward strand, and CpG SNPs using the reverse strand. The average amplicon length for these eight pairs was 372 bp (Table S3) to maximize the contiguous sequencing read length obtainable using the Ion S5 instrument (described below).
We performed polymerase chain reaction (PCR) on bisulphite-converted DNA in a total reaction volume of 10 μl using a standard 3-step PCR protocol (Table S4) and EpiMark Hot Start Taq DNA polymerase enzyme (New England Biolabs (NEB), Ipswich, Massachusetts). We visually compared the intensity and specificity of our PCR reactions using a 2% agarose gel stained with GelRed (Biotium, Fremont, California) and the quality and quantity of amplified, converted DNA was assessed using a NanoDrop UV/Vis spectrophotometer.
Quantitative polymerase chain reaction
As part of a prior study [Citation11], cDNA was generated from 500 ng of total RNA via reverse transcription by SuperScript III (Invitrogen, Carlsbad, CA), gene-specific primers, and oligo-dT. HTR2A expression was measured in triplicate at 25 different locations along the gene in all the samples by quantitative polymerase chain reaction (qPCR) using primers that are listed in Table [Citation5], as previously described [Citation20]. The different splice isoforms measured in this study were also previously described [Citation11], and include multiple exon 2 splice variants and the inclusion/exclusion of an extended 5'UTR. qPCR was performed on an ABI 7000 Sequence Detection System (Life Technologies) using 12.5 ng of cDNA per sample and Power SYBR Green PCR Master Mix (Life Technologies) in a 15 μl total reaction volume using. HTR2A expression was normalized to β-actin (ACTB) cycle threshold (CT) values.
In addition to qPCR analysis, we also measured the relative expression of exon 2 splice variants by performing PCR using a forward primer in exon 1 and a fluorescent-labelled reverse primer in exon 3. Transcript length and fluorescent intensity were resolved using an ABI 3730. The area under the curve (AUC) of the fluorescent peak corresponding to the amplicon length for each isoform (P n) was used as a measure of spliceoform expression level, measured in triplicate, and relative expression was calculated within each assay as individual AUC divided by the sum of the AUCs for all three isoforms, E2+, E2tr, and E2− (P n /ΣP 1 … P x).
Library preparation and sequencing
We sequenced the bisulphite-treated PCR amplicons using the Ion S5 Sequencing system (Ion Torrent, Carlsbad, CA). We prepared the sequencing libraries using the NEBNext Fast DNA Library Prep Set kit according to manufacturer instructions (NEB, Ipswich, MA). Briefly, blunt-ended fragments of the amplicons are repaired with a polymerase that ensures the two strands of each amplicon are the same length, allowing blunt-end ligation of sequencing adaptors containing a unique barcode for each sample. Ligation of unique barcodes allowed us to multiplex up to 96 samples per sequencing batch. Adaptor-ligated DNA was purified using .55X concentration of AMPure XP beads (Agilent, Santa Clara, California). We amplified the purified library for 6 PCR cycles using a high-fidelity polymerase. Finally, the library was cleaned using .55X concentration of AMPure XP beads. Barcoded samples were combined, templated onto IonSpheres, and loaded onto Ion 520 & 530 Chips using the Ion Chef. We used long read Ion ExT Chef reagents, to promote sequencing of 400–600 bp amplicons. We sequenced the loaded chips on the Ion S5 sequencer per the manufacturer’s instructions.
Bioinformatics and statistical analyses
Bioinformatics
All alignments use human reference genome build hg38. Sequenced reads from each library were mapped to the HTR2A gene locus with GSNAP [Citation21] using the “–use-’cmet’ command that permits mapping of bisulphite converted reads. We quantified methylation at each CpG site using the bcftools’ ‘pileup’ command for the aligned BAM files for each library. For allelic methylation analysis, we filtered contiguous reads that covered both rs6313 and adjacent CpGs using bedtools2. We then quantified the number of methylated and unmethylated cytosines on each allele of rs6313 in heterozygous samples.
Quantification of DNA methylation fraction and allelic methylation ratio
DNA methylation fraction at each CpG site /methylation-drug use.
For DNA methylation analysis, we selected HTR2A genomic regions encompassing common SNPs in the promoter and exon regions, where we anticipate that SNPs have a regulatory role in the expression of the gene and DNA methylation. We list these common SNPs in Table S1. We required at least one hundred reads at each CpG site for inclusion in our analyses. The percentage of methylation for each site was calculated by dividing the number of unconverted cytosines (i.e., methylated cytosines) by the total number of reads. In practice, this requires us to quantify the ‘C’ bases on the forward strand and the ‘G’ bases on the reverse strand, depending on which strand we targeted with our PCR primers.
Global DNA methylation associated with rs6311.
We measured the global contribution of r6311 genotype on the methylation status for all CpGs in the eight amplicons. We calculated the score for methylation for each rs6311 genotype by subtracting the percentage of methylation of each CpG in each homozygous (A/A, G/G) samples from the average of methylation for each CpG for all heterozygous (A/G) samples. We make the assumption in this analysis that heterozygous samples for rs6311 represent an intermediate methylation status relative to the homozygotes, such that if either of the homozygous genotypes deviates from the heterozygotes, it suggests a global effect of rs6311 on methylation. If no deviation from heterozygotes is observed, we conclude that rs6311 does not exert a global effect on methylation. For a detailed explanation, see the example below.
Score of CpG at chr13:46895624 for sample MB002 (A/A) = (percentage of methylation for CpG at chr13:46895624 for sample MB002 (A/A)) – (average of methylation for CpG at chr13:46895624 in all heterozygous sample (A/G))
Allelic methylation.
For allele-specific methylation analysis, we measured CpG methylation in a single amplicon containing rs6313 in 18 samples heterozygous for rs6313, which allows us to distinguish between the relative amount of methylation across the two alleles for rs6313. Heterozygosity at rs6313 enables us to distinguish between sequencing reads originating from each of the two alleles (A and G alleles), permitting quantification of relative methylation of the seven adjacent CpG sites across each allele contained in the amplicon. We calculated the DNA methylation percentage for each CpG in the eight amplicons. We performed allele-specific methylation (ASM) analysis for 18 individuals heterozygous for rs6313 (6 cocaine overdosed and 12 matched controls). We measured ASM in quadruplicate. We calculated the percentage of methylation for each CpG by counting the number of bisulphite-unconverted (C’s) and converted (T’s) across the A allele and G allele for rs6313 and then quantified the proportion per allele. The outcome is two methylation fraction values for each CpG; one for the A allele (minor allele methylation percentage) and one for the G allele (major allele methylation percentage). The ratio of these two fractions is the allelic methylation measure. Ratios greater than 1 indicate greater methylation on the minor (A) allele strand, while ratios less than 1 indicate greater methylation on the major (G) allele strand. An explanation follows.
Percentage of methylation CpG in minor allele strand = 100% * (counts of the number of C mapped at the reference C/ total number of reads) – A allele strand
Percentage of methylation CpG in major allele strand = 100% *(counts of the number of C mapped at the reference C/total number of reads) – G allele strand
Allelic ratio = Percentage of methylation CpG in minor A allele strand/Percentage of methylation CpG in major G allele strand
Statistical analysis
To minimize batch effects, we balanced the number of cocaine and control subjects in each sequencing batch, tested by using chi-square and t-test for all available covariates. These covariates included age, sex, race, smoking history, post-mortem interval (PMI), and RNA integrity number (RIN). To compare the distribution of clinical categorical variables (cocaine and control subjects, we used the chi-squared test (χ2) and Fisher exact test. We used independent samples t-tests to compare between-group distributions for continuous clinical variables (age, PMI, RIN). Demographic characteristics are available in Table S2.
We regressed the methylation percentage against the drug use status and the genotype of rs6311. We used multiple linear models for assessing the effect of drug use and the genotype of rs6311 on the methylation percentage of each CpG. We used Akaike Information Criterion (AIC) as a way to assess the quality of the model (model selection) and select covariates (age, race, sex, PMI, RIN) for each model. The corresponding analysis (e.g., ANOVA type III) assessed the statistical significance of all variables in each model. To control the rate of false-positive results for multiple testing (n tests = 51), we conservatively adjusted p values using Bonferroni correction. Significance level was set at 0.0009804 (Bonferroni corrected p-value).
For comparing the mean of the methylation score of each genotype of rs6311, we used the non-parametric Kruskal-Wallis rank-sum test and Dunn’s post-hoc test. For allelic-methylation analysis, we combined the reads of all four replicates for each sample according to the methylation status and allele type. We examined the association of methylation status with allele identity by χ2 goodness of fit. We calculated the expected values by dividing the total number of reads for each allele by two, assuming that methylation is uniformly distributed across each allele (i.e., the methylated percentage of each allele is 50%).
We performed Cochran Armitage test for trend to assess the association between the phenotype (cocaine addiction) and genotypes of following SNPs (rs1328685, rs6311, rs655888, rs2760351, rs6314, rs2246127, rs2070039, rs6313, rs6304, rs1328684, rs7330461, rs3803189, rs7324017, rs6312, rs76665058, rs73473857, rs61948307) using DescTools package in R [Citation22].
To assess the association between the genotypes of SNPs (rs1328685, rs6311, rs655888, rs2760351, rs6314, rs2246127, rs2070039, rs6313, rs6304, rs1328684, rs7330461, rs3803189, rs7324017, rs6312, rs76665058, rs73473857, rs61948307) and the methylation percentage of 51 CpGs sites in the eight amplicons we studied, we used the Matrix eQTL package in R [Citation23]. Matrix eQTL tested the association between genotypes of the previously mentioned SNPs and methylation percentage using linear regression assuming additive genotype effect. We included the following variables (age, ethnicity, sex, PMI, RIN, smoking history) as covariates. The relationship between methylation percentage and the expression was tested by Spearman correlation coefficient test. All statistical analyses were performed with R version 4.0.2.
Results
Cocaine has no significant association with the methylation level of individual CpG sites at the HTR2A locus
Bisulphite DNA sequencing of the HTR2A gene in 30 samples revealed no significantly differentially methylated CpG in cocaine users versus controls when controlling for multiple comparisons using Bonferroni-adjustment (Table S6).
rs6311 A/A genotype is associated with global hypermethylation of HTR2A
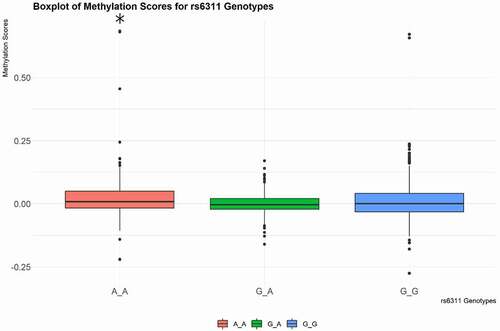
The methylation score across rs6311 homozygous genotypes significantly differed from heterozygous genotypes, suggesting a global effect of this SNP on CpG methylation across the HTR2A gene locus (Kruskal-Wallis rank-sum test, p-value = 0.006863), demonstrating that rs6311 was associated with overall greater methylation at the HTR2A locus. We performed Dunn’s Kruskal-Wallis post-hoc analysis to determine that A/A homozygotes had significantly higher methylation scores than G/G and A/G genotypes ().
Figure 1. Boxplot of the methylation score of rs6311 genotypes. The methylation score for A/A homozygotes was significantly higher than methylation scores for G/G and A/G genotypes (* p-value < .05).
Allelic methylation imbalance implicates genetic factors regulating HTR2A methylation
We performed a chi-square test to find if there is a significant difference in methylation between the two alleles, and then calculated the allelic ratio (see methods). Balanced allelic ratios (equally methylated on both alleles) would result in an allelic ratio score of 1. Samples significantly deviating from 1, based on p-value of the constructed chi-square test, would indicate imbalanced allelic methylation (i.e., one allele is significantly more methylated than the other). We examined allelic methylation ratios of seven CpG sites adjacent to rs6313. We excluded one sample due to the inadequate sequencing depth. Significant ratios are highlighted in green in Table S7. In summary, most of the significant allelic methylation ratios are larger than 1 (117 of 126 total calculated ratios), suggesting that the minor allele for rs6313 is hypermethylated. This finding is consistent with our global methylation findings showing the minor allele for rs6311 is hypermethylated, as rs6313 and rs6311 are in near perfect linkage disequilibrium (LD).
No significant correlation between methylation at the level of individual CpG sites and SNP Genotype
Using Matrix eQTL, we analysed the SNP-CpG associations across the genotypes of 17 SNPs (rs1328685, rs6311, rs655888, rs2760351, rs6314, rs2246127, rs2070039, rs6313, rs6304, rs1328684, rs7330461, rs3803189, rs7324017, rs6312, rs76665058, rs73473857, rs61948307) and the methylation of all 51 CpG sites. Based on the minimum FDR values across all eQTLs, there is no significant correlation between the genotype of the SNPs and the DNA methylation level of individual CpG dinucleotides after adjusting for multiple comparisons (Table S8).
(A) allele of rs6311 is a risk allele for cocaine addiction (Association of genotype of rs6311 of HTR2A gene and risk of cocaine addiction)
We used the Cochran-Armitage test as the distribution of genotypes rs6311 (Table S9 & S10) in the population deviates from H-W proportion. We used the Cochran–Armitage test to assess if the proportion of cocaine addicts is different for genotypes of rs6311. The results showed linearity decreasing trend between drug use status and the number of G alleles for rs6311 (p-value = 0.04156 (two-sided test).
rs6311 and rs6313 both have indirect role in modulating the expression of exon 2 in HTR2A
We tested if there is a significant association between the expression of HTR2A and the genotype of following SNPs (rs1328685, rs6311, rs655888, rs2760351, rs6314, rs2246127, rs2070039, rs6313, rs6304, rs1328684, rs7330461, rs3803189, rs7324017, rs6312, rs76665058, rs73473857, rs61948307). Among all the previous SNPs, only rs6311 and rs6313 showed a significant differential expression of HTR2A gene. The minor alleles for both rs6313 and rs6311 SNPs are associated with significantly higher relative expression of truncated exon 2 splice isoforms (Table S11).
No significant correlation between the methylation at the level of individual CpG sites and the expression of HTR2A transcripts
In order to test whether there is an association between the percentage of methylation at any of the CpG sites and the expression of various HTR2A transcripts, we used Spearman’s rank correlation coefficient as both methylation and expression data sets are not normally distributed. We found associations between the methylation of some of CpGs sites and the expression of transcripts, but none of these of the associations survived correction for multiple testing using ‘Holm’s method for correcting for multiple inferences.
Cocaine exposure associated significantly with the expression of exon 2 truncated splice isoform and the total expression of HTR2A gene
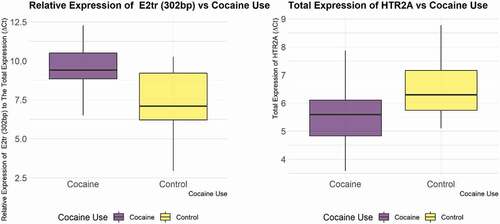
We regressed the threshold value of all the mentioned transcripts we studied against the drug use status, adjusting for the genotypes of rs6311. We used linear models for assessing the effect of drug use and the genotype of rs6311 on the expression of each one of the transcripts. We used Akaike Information Criterion (AIC) to assess the quality of the model (model selection) and select covariates (age, race, sex, PMI, RIN, smoking history) for each model. The corresponding analysis (e.g., ANOVA type III) assessed the statistical significance of all variables in each model. Significance level was set at p < 0.05. Cocaine addiction showed a significant association with the total expression of HTR2A gene (total expression of HTR2A expression) (p-value for drug use (cocaine) = 0.03668) and the relative expression of truncated exon 2 splice isoform (p-value for drug use = 0.008092). Cocaine is significantly associated with higher total expression of HTR2A but with lower expression of truncated exon 2 isoform ().
Figure 2. Relative expression of truncated exon 2 splice isoform to total HTR2A expression in cocaine group vs. control group (left panel) and the total expression of HTR2A in the cocaine group vs. control group, normalized to β-actin. Note: higher Ct values indicate lower expression.
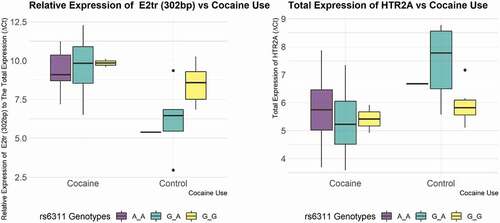
Figure 3. Relative expression of truncated exon 2 splice isoform to total HTR2A expression in cocaine and control groups with respect to rs6311 genotypes (left panel) the total expression of HTR2A in cocaine group vs. control group with respect to rs6311 genotypes, normalized to β-actin. Note: higher Ct values indicate lower expression.
Discussion
Cocaine dependence is a complex disease where addiction susceptibility in individuals is the result of the interaction of environment and genetic background [Citation24]. This interaction is regulated at the molecular level by epigenetic mechanisms. CpG methylation is a major epigenetic mechanism that regulates DNA replication, translation, and mRNA expression [Citation11]. However, the methylation status of HTR2A and its role in modulating the expression of HTR2A was unknown in the context of cocaine addiction. Therefore, revealing this complex interaction between environmental exposure, epigenetic modification, and genetic variants is an important first step towards understanding the aetiology of cocaine use disorder as a complex trait. In this study, we revealed the interaction between epigenetic modifications, specifically DNA methylation, and genetic variants in HTR2A in the context of cocaine addiction. We achieved this by 1) Studying DNA methylation pattern of CpGs sites in HTR2A in overdosed cocaine subjects and their matched controls, 2) Identifying the global effect of rs6311 genotypes on the methylation pattern in HTR2A, 3) Identifying the allele-specific DNA methylation patterns for rs6313 in HTR2A, and 4) studying HTR2A gene expression profiling and its association to methylation and genetic variants in overdosed cocaine subjects and their matched control.
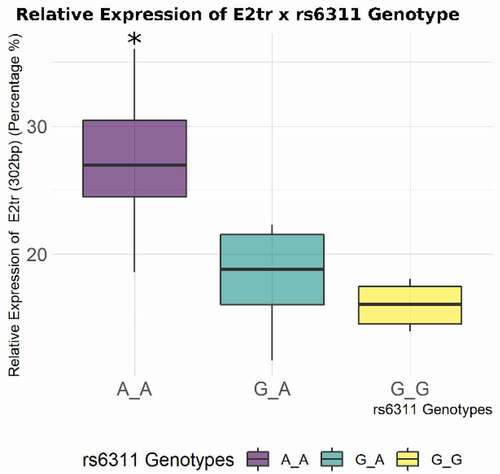
Our study of HTR2A gene expression and methylation in context of addiction revealed a number of complex associations between variants, expression, and the methylation of HTR2A gene. Among all the SNPs that we tested for genetic association to cocaine addiction, rs6311 (and rs6313, based on LD) showed a significant allelic association in cocaine overdose subjects, whereby the A/A genotype of rs6311 is associated with cocaine use. However, this analysis is severely underpowered and needs additional follow-up analysis in a larger population. In term of methylation, we also found the A/A genotype of r6311 has a global effect on the methylation status of the HTR2A gene. This is consistent with previous data that showed the A/A genotype had a higher CpG methylation percentage at a cytosine located in exon 2 (chr13:47469654; GRCh37/hg19), when compared to homozygous G/G major allele carries [Citation11]. This finding lends further support for the regulatory role that this SNP plays in the methylation of HTR2A. Moreover, our study showed that the homozygous A/A genotype rs6311 is associated with the higher relative expression of the truncated exon 2 splice isoform . Previous research showed that rs6311 A/A genotype decreases expression for an mRNA isoform of HTR2A encoding a long 5ʹUTR [Citation25]. Cocaine use was also associated with lower relative expression of the splice isoform in which exon 2 is truncated, and with higher total expression of HTR2A. In summary, both rs6311 and cocaine are associated significantly with expression of the truncated exon 2 splice isoform. In addition, the A/A genotype of rs6311, which is associated with higher global methylation of HTR2A, is associated with cocaine use.
Figure 4. Relative percentage of truncated exon 2 splice isoform (as estimated by AUC), versus E2+ and E2− isoforms with respect to rs6311 genotypes. A/A homozygotes express significantly more of the E2tr isoform relative to G/G homozygotes and heterozygotes (*p < 0.05).
Given this association between A/A rs6311 genotype, higher global methylation of HTR2A gene and cocaine use, and the association between relative expression of truncated exon 2 splice isoform and rs6311 and cocaine use, we find it possible that this genotype contributes to cocaine abuse liability by increasing methylation across this locus, affecting mRNA expression levels, thereby altering 5-HT signalling through the 5-HT2A receptor. Thus, rs6311/A is a potential risk allele that can help us understand the role of 5-HT signalling in cocaine addiction and could serve as a biomarker for hypermethylation at the HTR2A locus in the human brain. rs6311/A would represent an allele with reduced signalling, as meta-analysis of selective serotonin reuptake inhibitors (SSRIs) drug response showed the A/A genotype is associated with fewer side effects of SSRIs, and less response to SSRIs treatment [Citation26], consistent with reduced 5-HT2A signalling.
Allele-specific methylation analysis showed that the A allele of rs6313, which corresponds to the A allele of rs6311, has higher methylation compared to G allele for the CpGs adjacent to rs6313. Imbalanced allelic methylation strongly suggests the presence of genetic factors modulating methylation differently across the two alleles. We speculate that methylation of the A allele impacts HTR2A transcription by inhibiting the recruitment of transcription factors that bind to the methylated CpGs. Importantly, we found significant but modest differences in allelic methylation, suggesting a direct genetic effect on DNA methylation. The patterns of allelic methylation across CpG sites are not the same across each sample. We interpret this finding in a number of ways. First, we conclude that the actual SNP affecting DNA methylation is not likely to be rs6313, but another SNP (or group of SNPs) that affect methylation in cis. It is possible that a combination of SNPs (haplotype) is associated with specific DNA methylation patterns, finely tuning methylation in this region. It is also possible that the magnitude of allelic methylation differences could be influenced by environmental factors, such that the allele is primed for greater methylation following specific (unknown) environmental exposures. However, none of the SNPs studied, including rs6311 and rs6313, showed a significant correlation with the methylation at the level of individual CpG dinucleotides after adjusting for multiple comparisons. Perhaps, the combined effect of changes in the methylation governs the changes in the expression and consequently developing the phenotype rather than the change in methylation at the level at one CpG site.
The association of rs6311 (and rs6313, based on LD), HTR2A expression, and methylation has been investigated in several studies. C-allele carriers of rs6313 (G-allele of rs6311) showed lower methylation at one CpG site located 1.2 kb upstream of rs6313as compared to the T/T genotype rs6313 [Citation27]. One study investigating the methylation changes in the blood samples of cocaine subjects showed that the two CpG site at the promoter region of HTR2A is hypomethylated in cocaine users vs. non-users. The methylation of these two CpG sites has also been associated with cocaine relapse behaviour [Citation28]. However, in our study, we did not find any significant difference in the methylation at the level of one CpG site between the healthy control subjects and cocaine subjects. It is possible that our study sample was not large enough to detect this modest change. In addition, DNA methylation is a dynamic tissue-specific phenomenon, whereby changes in the blood might not correspond to changes in brain tissues.
In another study that examined DNA methylation association with rs6311 and stress in preschool children, children homozygous rs6311 for (A/A) had lower mean methylation of HTR2A at the two CpG sites in the promoter region (−1420 and −1224). Furthermore, stress was positively correlated with methylation in A/A genotype subjects, while negatively correlated with methylation among GG subjects [Citation19]. Again, this observation, which differs from the current study, could be explained by the saliva samples being used to detect the methylation changes, and the difference in study cohorts, in terms of age and disease/phenotype conditions.
In previous studies, the promoter region of HTR2A showed different methylation patterns depending on the region’s location and the genotype of rs6311 and rs6313. Compared to control subjects, the promoter region was hypomethylated near rs6313 vs. hypermethylated around rs6311 in bipolar disorder and schizophrenia subjects [Citation29]. In the present study, our DNA methylation analysis did not show any difference in the methylation in the promoter region according to the genotype of rs6311 (rs6313). Abdolmaleky’s group also found that A allele-rs6311 carrying subjects showed lower expression of the HTR2A gene compared to the GG genotype subjects [Citation29]. In the current study, the AA genotype showed lower total expression of the HTR2A gene among cocaine subjects (, left panel).
The discrepancies between the current and previous studies are possibly explained by several factors: differences in the sample source used (blood, saliva, vs. post-mortem brain tissue) and the age of the ‘study’s subjects, as some of the studies mentioned above, have been conducted on children and toddlers. In contrast, the current study’s subjects are adults with an average age of 41 years old. Furthermore, methylation has been examined in the context of different diseases. Given that methylation differs according to the tissue, age, diseases status, and environmental factors, the results of the present study specifically examined DNA methylation in post-mortem tissue of adult subjects with and without a history of cocaine addiction. These differences underline the importance of studying DNA methylation in the proper context.
Several limitations of the current study should be pointed out. First, we examined allelic methylation for only one SNP. Although we found a statistically significant deviation of allelic ratios for rs6313, the ratios were not consistent across the samples, precluding a clear conclusion about which SNP (or SNPs) are directly influencing methylation. However, this finding lends strong support for a direct genetic effect on CpG methylation at this locus. Second, the allelic methylation analysis of rs6313 has been conducted in heterozygous samples in regardless of their cocaine addiction status due to the small size sample. However, the allelic methylation analysis results might be different in cases versus controls. Therefore, this analysis needs to be replicated in larger sample sizes and taking into account the cocaine addiction status in consideration. Third, this study lacks statistical power due to the low sample size, which reduces the chance of detecting an actual effect from false-positive results, requiring further replication with large sample sizes to confirm these preliminary results. Even with these limitations, our observations converge, leading us to conclude that rs6311 and rs6313 modulate methylation and expression at the HTR2A gene locus in the context of cocaine addiction.
Conclusion
This study illustrates that rs6311 and rs6313 play a role in regulating HTR2A by modulating expression and methylation. Specifically, rs6311 A/A genotype appears to increase global methylation at the HTR2A gene locus. This effect is consistent with imbalanced allele-specific methylation, where we observed higher methylation of the A allele relative to the G allele. We were also able to illustrate the allelic association of r6311 with cocaine addiction, where we observed a higher frequency of cocaine addicts associated with increasing the dose of A allele of rs6311 (as different frequency between cases and control with respect to the average number of A allele of rs6311). These findings add further validation for 5-HT2A’s involvement in cocaine addiction. rs6311 appears to alter the methylation status and the expression of HTR2A, which consequently modulates the function and the signalling of 5-HT2A receptor thereby influencing the variation in the risk for cocaine addiction.
Supplemental Material
Download MS Word (40.8 KB)Acknowledgments
The authors sincerely thank the tissue donors and their next of kin for donating the brain tissues used in this study. The authors also thank the laboratories of Dr. Wolfgang Sadee and Dr. Deborah Mash for their scientific expertise and contributions in curating these tissues.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- National Institute on Drug Abuse. Costs of substance abuse. 2015. Available from: https://archives.drugabuse.gov/trends-statistics/costs-substance-abuse
- What is the scope of cocaine use in the United States? [Internet]. 2016. Available from: https://www.drugabuse.gov/publications/research-reports/cocaine/what-scope-cocaine-use-in-united-states
- Nestler EJ. The neurobiology of cocaine addiction. Science & practice perspectives/a publication of the National Institute on Drug Abuse, National Institutes of Health; 2005;3:4–10.
- Sofuoglu M, Sewell RA. Norepinephrine and stimulant addiction. Addict Biol. 2009;14(2):119–129.
- Filip M, Frankowska M, Zaniewska M, et al. The serotonergic system and its role in cocaine addiction. Pharmacological Reports; 2005;57:685–700.
- Paquette AG, Marsit CJ. The developmental basis of epigenetic regulation of HTR2A and psychiatric outcomes. J Cell Biochem. 2014;115(12):2065–2072.
- Keys Lawler AC, Torres-Yaghi Y, Amjad F, et al. Reader response: pimavanserin: a novel therapeutic option for Parkinson disease psychosis. Neurol Clin Pract. 2018;8(3):175.
- Fletcher PJ, Phil D, Grottick AJ, et al. Differential Effects of the 5-HT 2A Receptor Antagonist M100, 907 and the 5-HT 2C Receptor Antagonist SB242, 084 on Cocaine-induced Locomotor Activity, Cocaine Self-administration and Cocaine-induced Reinstatement of Responding. 2002;2.
- Cao J, Liu X, Han S, et al. Association of the HTR2A gene with alcohol and heroin abuse. Hum Genet. 2014;133(3):357–365.
- Ruble CL, Smith RM, Calley J, et al. Genomic structure and expression of the human serotonin 2A receptor gene (HTR2A) locus : identification of novel HTR2A and antisense (HTR2A-AS1) exons. BMC Genet. 2016;17(Suppl 2):1–15.
- Smith RM, Papp AC, Webb A, et al. Multiple regulatory variants modulate expression of 5-hydroxytryptamine 2A receptors in human cortex. Biol Psychiatry. 2013;73(6):546–554.
- HTR2A - 5-hydroxytryptamine receptor 2A - Homo sapiens (Human) [Internet]. HTR2A gene & protein. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information; 2004. Available from: https://www.ncbi.nlm.nih.gov/gene/3356
- Fernàndez-Castillo N, Roncero C, Grau-Lopez L, et al. Association study of 37 genes related to serotonin and dopamine neurotransmission and neurotrophic factors in cocaine dependence. Genes, Brain Behav. 2013;12(1):39–46.
- Filip M, Bubar MJ, Cunningham KA. Contribution of serotonin (5-hydroxytryptamine; 5-HT) 5-HT2 receptor subtypes to the hyperlocomotor effects of cocaine: acute and chronic pharmacological analyses. J Pharmacol Exp Ther. 2004;310(3):1246–1254.
- Pockros LA, Pentkowski NS, Swinford SE, et al. Blockade of 5-HT2A receptors in the medial prefrontal cortex attenuates reinstatement of cue-elicited cocaine-seeking behavior in rats. Psychopharmacology (Berl). 2011;213(2–3):307–320.
- Nic Dhonnchadha BA, Fox RG, Stutz SJ, et al. Blockade of the serotonin 5-HT2A receptor suppresses cue-evoked reinstatement of cocaine-seeking behavior in a rat self-administration model. Behav Neurosci. Internet]. 2009;123(2):382–396.
- Paquette AG, Lesseur C, Armstrong DA, et al. Placental HTR2A methylation is associated with infant neurobehavioral outcomes. Epigenetics. 2013;8(8):796–801.
- Ghadirivasfi M, Nohesara S, Ahmadkhaniha H-R, et al. Hypomethylation of the serotonin receptor type-2A Gene (HTR2A) at T102C polymorphic site in DNA derived from the saliva of patients with schizophrenia and bipolar disorder. Am J Med Genet Part B Neuropsychiatr Genet. 2011;156(5):536–545.
- Parade SH, Novick AM, Parent J, et al. Stress exposure and psychopathology alter methylation of the serotonin receptor 2A (HTR2A) gene in preschoolers. Dev Psychopathol. 2017;29(5):1619–1626.
- Smith RM, Alachkar H, Papp AC, et al. Nicotinic α5 receptor subunit mRNA expression is associated with distant 5' upstream polymorphisms. Eur J Hum Genet. 2011;19(1):76–83.
- Wu TD, Fast NS. and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26(7):873–881.
- Signorell A, Aho K, Alfons A, et al. {DescTools}: tools for Descriptive Statistics. 2021. Available from: https://cran.r-project.org/package=DescTools
- Shabalin AA. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012;28(10):1353–1358.
- Pierce RC, Fant B, Swinford-Jackson SE, et al. Environmental, genetic and epigenetic contributions to cocaine addiction. Vol. 43. Neuropsychopharmacology. Springer US; 2018. p. 1471–1480.
- Smith RM, Banks W, Hansen E, et al. Family-based clinical associations and functional characterization of the serotonin 2A receptor gene (HTR2A) in autism spectrum disorder. Autism Res. 2014;7(4):459–467.
- Kato M, Serretti A. Review and meta-analysis of antidepressant pharmacogenetic findings in major depressive disorder. Mol Psychiatry. 2010;15(5):473–500.
- Cheah S-Y, Lawford BR, Young RM, et al. mRNA expression and DNA methylation analysis of serotonin receptor 2A (HTR2A) in the human schizophrenic brain. Genes (Basel). 2017;8(1):1.
- Land MA, Ramesh D, Miller AL, et al. Methylation Patterns of the HTR2A associate with relapse-related behaviors in cocaine-dependent participants. Front Psychiatry. 2020;11:532.
- Abdolmaleky HM, Yaqubi S, Papageorgis P, et al. Epigenetic dysregulation of HTR2A in the brain of patients with schizophrenia and bipolar disorder. Schizophr Res. 2011;129(2–3):183–190.