
ABSTRACT
Genome methylation profiles define naïve-like (n-CLL), memory-like (m-CLL), and intermediate (i-CLL) subsets of chronic lymphocytic leukaemia (CLL). The profiles can be easily determined by the analysis of the five-CpG signature. m-CLL, i-CLL, and n-CLL with the good, intermediate, and poor prognoses, respectively, differ by the somatic hypermutation status of the immunoglobulin heavy chain variable gene (IGHV), a widely used prognostic predictor in CLL. We have previously shown that the expression of WNT5A, encoding a ROR1 ligand, distinguishes patients with the worse outcome within the prognostically favourable IGHV-mutated subgroup. To analyse the mechanisms controlling WNT5A expression, we investigated the methylation status of 54 CpG sites within the WNT5A promoter and its relation to the WNT5A gene expression. In a cohort of 59 CLL patients balanced for combinations of IGHV and WNT5A statuses, we identified three promoter CpG sites whose methylation level correlated with the WNT5A expression within the IGHV-mutated subgroup. Further, we complemented our data with the methylation status of the five-CpG signature. IGHV-mutated/WNT5A-negative and IGHV-mutated/WNT5A-positive cases overlapped with m‑CLL and i‑CLL methylation subgroups, respectively, while most IGHV‑unmutated samples were assigned to n-CLL. Median methylation levels of all the three CpG sites in the WNT5A promoter were lowest in i-CLL. Finally, a detailed analysis of m-CLL and i-CLL showed that undetectable WNT5A expression predicts longer treatment-free survival with higher statistical significance than the classification according to the five-CpG signature. To conclude, a favourable m-CLL subgroup is associated with mutated IGHV and undetectable WNT5A expression due to its promoter methylation.
Background
DNA methylation plays a vital role during the maturation of B-cells, and it is crucial for their normal functioning [Citation1–3]. During the maturation process, DNA undergoes global hypomethylation with local hypermethylation, and a similarly occurring process is even more pronounced in cells of chronic lymphocytic leukaemia (CLL) [Citation3,Citation4]. Aberrant changes in the methylation profile of CLL cells accumulate during early leukaemogenesis; afterwards, the overall profile remains more or less stable [Citation1,Citation3–6]. The methylation status of the five-CpG signature was shown to divide patients into subgroups based on their similarity to B-cell developmental stages [Citation6]: (i) Naïve-like CLL (n-CLL) with a high methylation level, associated with a poor prognosis of the patients, (ii) memory-like CLL (m-CLL) with a low methylation level, associated with a good prognosis, and (iii) intermediate-CLL (i-CLL) with both methylation and prognosis in-between [Citation6,Citation7]. These three methylation subgroups partially overlap with prognostic categories defined by the somatic hypermutation status of the immunoglobulin heavy chain variable gene (IGHV), an important prognostic predictor [Citation8–11]. CLL patients with IGHV identity to germline ≥98% (IGHV-unmutated) have a poor prognosis. However, even within the prognostically favourable IGHV-mutated subgroup, there is a subset of patients with an aggressive disease course [Citation12,Citation13].
We have previously shown that the expression of WNT5A, a gene encoding a ligand activating WNT/Planar Cell Polarity pathway via the ROR1 receptor, varies significantly among CLL patients, from high levels in some patients to undetectable levels in 50% of IGHV-unmutated and 85% of IGHV-mutated cases (marked as WNT5A-negative) [Citation13]. WNT5A expression is a strong and overtime-stable prognostic predictor, distinguishing cases with worse prognosis within otherwise favourable IGHV-mutated CLL better than the percentage of IGHV identity [Citation12,Citation13].
Considering the absence of WNT5A expression in more than half of tested patients [Citation13], together with reports of a correlation between the WNT5A expression and its promoter methylation in other cancer types [Citation14–16], we hypothesized that the WNT5A expression might be regulated by methylation also in CLL. Further, taking advantage of the current understanding of the CLL methylation profiles, we explored how methylation within the WNT5A promoter correlates with the global methylation profile, IGHV status, and WNT5A expression.
Material and methods
Patient samples
CLL cells were isolated from peripheral blood of 59 CLL patients monitored and treated at the University Hospital Brno. All samples were obtained after written informed consent in concordance with the Declaration of Helsinki, and the study was approved by the Ethical Committee of the University Hospital Brno. B-cells from patient samples were separated using B-cell Enrichment RosetteSep kits (StemCell Technologies). The original cohort consisted of 39 patients; another 20 IGHV-mutated patients were added to refine the results. The assessment of WNT5A expression and IGHV gene mutational status were processed as previously described [Citation13].
DNA isolation and bisulphite conversion
DNA was isolated on QIAcube (Qiagen) and dissolved in TE buffer with 0.1 M EDTA. DNA (200 ng) was treated with an EZ DNA MethylationTM kit (Zymo Research) according to the manufacturer’s recommendation and eluted into 22 μl of water.
Primer design for WNT5A promoter analysis
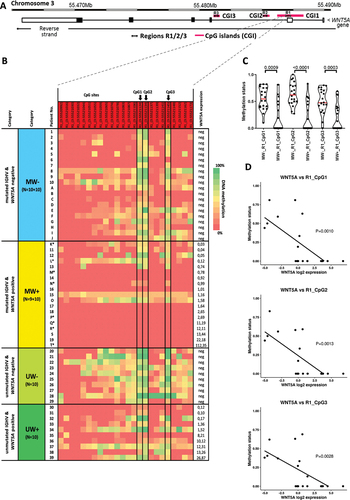
The sequences of three CpG Islands (CGI1–CGI3) located within the WNT5A promoter (Ensembl databases, version GRCh37.p13) were used for primer design. The sequences were modified to match bisulphite-converted DNA. The Primer3 tool (http://bioinfo.ut.ee/primer3-0.4.0/) was used for primer design. The parameters of the primers were tested with OligoAnalyzer 3.1 (https://eu.idtdna.com/calc/analyser) and ePCR (http://bisearch.enzim.hu/) tools. Primers were synthesized by Generi Biotech (Table S1). The location of the examined regions is illustrated in the scheme in .
Figure 1. Methylation analysis of WNT5A promoter. (a) Schematic location of the regions R1, R2, R3 amplified with PCR within the CpG Islands CGI1/2/3 of the WNT5A promoter. (b) Heatmap displaying the results of the methylation analysis of the region R1 within CpG Island CGI3 in the total cohort of 59 CLL patients divided into four groups based on IGHV status and WNT5A expression. The initial cohort of 39 patients (described with numbers; see Figure S1 for initial analysis of regions R1, R2 and R3), was extended with 10 patients per MW- and MW+ cohorts each (described with letters). Arrows point to the three CpG sites R1_(3:55,521,124), R1_(3:55,521,134), and R1_(3:55,521,145), the methylation status of which correlated with the WNT5A expression in the IGHV-mutated subset. *Sample taken in relapse after treatment. (c) Comparison of the methylation level between MW- and MW+ samples for all three CpG sites (Mann–Whitney test). (d) The negative correlation between the WNT5A expression and methylation status of the three CpG sites (Spearman correlation) within the IGHV-mutated subset.
PCR and sequencing
Bisulphite-treated DNA was amplified with PCR using HotStarTaq DNA Polymerase (Qiagen) in reaction conditions: 5 min 95°C, (30 sec 95°C, 30 sec 60°C, 1 min 72°C) x 35 cycles, 7 min 72°C. PCR products were sequenced either by GATC Biotech or in-house using ABI PRISM® 3700 Genetic Analyser according to the manufacturer’s instructions (Big Dye terminator v1.1 kit, Applied Biosystems). Primer sequences for the WNT5A promoter regions are listed in Table S1; primers for the five-CpG signature analysis were used as published previously [Citation6].
Statistical analyses
The following tests were used to verify distribution normality: Kolmogorov–Smirnov test, Shapiro–Wilk test, or D´Agostino-Pearson normality test. Parametric and non-parametric tests (for normally and non-normally distributed variables, respectively) were used to evaluate the relationships between variables (unpaired t-test and Mann Whitney test) and correlation between two variables (Pearson and Spearman correlation). Differences in survival were analysed by the log-rank test. The level of statistical significance was set to P < 0.05. In multiple testing, the P-value was adjusted using the Holm–Bonferroni method. All assays were performed as two-tailed using GraphPad Prism 8 (GraphPad Software Inc., La Jolla, CA, USA) and R program (http://www.r-project.org; univariate and multivariate survival analysis). Within TFS, treatment or death due to CLL was treated as an event.
Results:
Using ENSEMBL GRCh37, we identified 54 CpG sites in three regions within the WNT5A promoter localized in three CpG Islands (CGI1–CGI3): region 1 (R1) in CGI1, region 2 (R2) in CGI2, and region 3 (R3) in CGI3 (). For the initial experiment, 39 previously untreated CLL patients were classified based on IGHV status and WNT5A expression: IGHV-mutated/ WNT5A-negative (MW-), IGHV-mutated/WNT5A-positive (MW+), IGHV-unmutated/WNT5A-negative (UW-), IGHV-unmutated/WNT5A-positive (UW+) (Figure S1 and numbered samples in ). In separated B-cells, we explored the methylation status of the 54 individual CpG sites using bisulphite conversion (EZ DNA Methylation kit; Zymo Research) followed by Sanger sequencing. We used a more precise approach that provides information about the methylation level of individual CpG sites compared to other studies using methylation-specific PCR, providing only the overall methylation status of the whole measured locus [Citation14–17]. We identified three CpG sites within R1 (further indicated as R1_CpG1, R1_CpG2, and R1_CpG3), whose methylation levels negatively correlated with WNT5A expression within the IGHV-mutated subset. We detected high methylation levels of these three CpG sites in MW-patients, while in MW+ patients, the methylation level decreased with increasing WNT5A expression. We extended the cohort of IGHV-mutated patients (10 MW- and 10 MW+; ) and confirmed significant differences in methylation of these three CpG sites (; R1_CpG1: P = 0.0014, R1_CpG2: P = 0.0001, R1_CpG3: P = 0.0005; Mann–Whitney test). We also confirmed the negative correlation between WNT5A methylation and expression (MW+ cohort; : R1_CpG1: P = 0.001, R1_CpG2: P = 0.0013, R1_CpG3: P = 0.0028; Spearman correlation).
We observed high methylation levels in UW-patients with a median methylation level being even higher than in MW-patients ( and S2A). In contrast, we have not seen any specific dependency between WNT5A methylation and the expression levels in UW+ patients (Figure S2B). It implies that the WNT5A expression in the IGHV-unmutated subset is driven by different mechanisms than mere WNT5A promoter methylation.
Furthermore, when we compared our classification based on the combination of IGHV status and WNT5A expression to the classification based on the five-CpG signature described by Queirós et al. [Citation6], we found that both largely overlapped. The two prognostically distinct IGHV-mutated subgroups, MW- and MW+ that differed by WNT5A promoter methylation and WNT5A expression, corresponded to the memory-like (m-CLL) and intermediate (i-CLL) methylation subgroups, respectively (). In contrast, except for two patients belonging to i-CLL, all IGHV-unmutated patients from our cohort were assigned to the naïve-like CLL methylation subgroup. In agreement with published data [Citation6], we detected significant differences in the IGHV somatic hypermutation load. All n-CLL patients had IGHV identity above 99.1%, the two i-CLL IGHV-unmutated cases had identity below 99%, and the rest of i-CLL (18 of 20) and all m-CLL were IGHV-mutated with identity below 98%; i-CLL patients had mostly borderline IGHV identity (). The WNT5A promoter methylation levels also significantly varied among individual methylation subgroups: m-CLL vs. i-CLL (P < 0.0001) and m-CLL vs. n-CLL (P = 0.0183) (). This data suggests that hypomethylation of CpG regions in the WNT5A promoter largely overlaps with i-CLL.
Figure 2. CLL patient classification into three methylation subgroups n-CLL, i-CLL, m-CLL, and treatment-free survival. (a) CLL patients were classified into the three subgroups based on the methylation profile of the five-CpG signature identified by Queirós et al. [Citation6]. The three methylation subgroups were further investigated for the association with IGHV identity (b) and with the methylation levels of the three CpG sites in the WNT5A promoter (c-e). Treatment free-survival prediction in the cohort of m-CLL and i-CLL subgroups (N = 41 patients) according to the WNT5A expression (f) and the five-CpG signature (g).
![Figure 2. CLL patient classification into three methylation subgroups n-CLL, i-CLL, m-CLL, and treatment-free survival. (a) CLL patients were classified into the three subgroups based on the methylation profile of the five-CpG signature identified by Queirós et al. [Citation6]. The three methylation subgroups were further investigated for the association with IGHV identity (b) and with the methylation levels of the three CpG sites in the WNT5A promoter (c-e). Treatment free-survival prediction in the cohort of m-CLL and i-CLL subgroups (N = 41 patients) according to the WNT5A expression (f) and the five-CpG signature (g).](/cms/asset/82fc60b5-3ffd-4cee-aae7-a1c3dbf9fcbe/kepi_a_2050004_f0002_oc.jpg)
Since the vast majority of m-CLL and i-CLL corresponded to IGHV-mutated CLL, we also decided to analyse treatment-free survival (TFS) and overall survival (OS) based on the WNT5A expression in the cohort consisting of m-CLL and i-CLL subgroups. Both the WNT5A-positivity () and classification into i-CLL methylation subgroup () distinguished patients with shorter TFS; the WNT5A positivity was a stronger prognostic factor than global epigenetics (i-CLL vs. m-CLL) (P = 0.0009 vs. P = 0.0311). Neither classification system showed a significant difference in OS for these subgroups of patients (Figure S3).
Discussion
The Wnt5a ligand signals via the ROR1 receptor that is highly expressed on CLL cells [Citation18–20]. The Wnt5a/ROR1 axis has been shown to regulate multiple aspects of CLL biology – including cell survival, migration, and proliferation [Citation13,Citation21–25]. Importantly, a high activity of the non-canonical Wnt pathway has been connected to the poor outcome of CLL patients [Citation13,Citation21,Citation26]. Following our previous work, here we verified that the WNT5A expression is a robust prognostic marker distinguishing CLL patients with worse prognosis within the prognostically favourable subset of IGHV-mutated patients.
In CLL, multiple epigenetic changes have been associated with the Wnt pathway (for review, see [Citation27]). Interestingly, there is no direct evidence for epigenetic silencing of ‘activators’ such as WNT ligands and FZD receptors. This contrasts with Wnt pathway inhibitors such as members of the SFRP family or WIF1 that have been found methylated in CLL [Citation28–31]. Our study thus represents the first observation of the epigenetic control of a Wnt ligand in CLL as we demonstrated that the WNT5A expression is associated with DNA methylation changes of WNT5A promoter in a significant proportion of CLL patients. The methylation status of three CpG sites within the WNT5A promoter correlated with WNT5A expression only in the IGHV-mutated but not in the IGHV-unmutated subgroup, suggesting that WNT5A expression in these two CLL subgroups is controlled via different mechanisms.
Furthermore, we confirmed that the five-CpG signature divides patients into the three prognostically distinct subgroups: m-CLL, i-CLL, and n-CLL [Citation6,Citation7]. The i-CLL epitype had been poorly characterized until recently when it was linked to specific biological and clinical features, namely the usage of IGLV3-21R110 [Citation32,Citation33], stereotyped BCR immunoglobulins (mainly of subset #2), increased frequency of SF3B1 and ATM mutations, and unfavourable prognosis [Citation34,Citation35]. Very recently published paper recognized WNT5A/B overexpression as a specific signature of patients carrying IGLV3-21R110 [Citation35]; this corresponds to our results and our previous work showing that among IGHV-mutated patients, the WNT5A expression is high in patients with borderline number of IGHV somatic hypermutations and SF3B1 mutations and more aggressive disease compared to WNT5A-negative patients [Citation13].
While the majority of patients with unmutated IGHV from our cohort were assigned to n-CLL, the WNT5A expression and methylation status of patients with mutated IGHV was strongly related to their distribution between the m-CLL and i-CLL subgroups. In our study, the classification based on the WNT5A expression distinguished patients with shorter TFS with even a higher significance than their classification into the m-CLL and i-CLL subgroups defined by the five-CpG signature.
To summarize, the level of WNT5A expression has been associated with CLL clinical behaviour [Citation13,Citation35]. Our current findings shed light on the interconnection of the varying WNT5A expression with the methylation profile of IGHV-mutated patients, prevalently comprising the m-CLL and i-CLL subsets as based on the five-CpG signature. While the methylated CpG sites in the WNT5A promoter of the m-CLL subtype patients correlated with undetectable WNT5A expression, the demethylation of these WTN5A promoter sites was shown in the i-CLL subset and associated with the high WNT5A expression. Together with previously published observations, our present results could serve for future designing better stratification models distinguishing patients with different prognoses.
Supplemental Material
Download MS Power Point (836.8 KB)Acknowledgments
The authors thank Francesco Muto for language editing and Lenka Radova and Karol Pal for help with figures. The internship of L.P. in Barcelona was funded by the Erasmus+ Programme of the European Union. V.B. and P.J. were supported by European Structural and Investment Funds, Operational Programme Research, Development and Education– “Preclinical Progression of New Organic Compounds with Targeted Biological Activity” (Preclinprogress) - CZ.02.1.01/0.0/0.0/16_025/0007381.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
References
- Oakes CC, Seifert M, Assenov Y, et al. DNA methylation dynamics during B cell maturation underlie a continuum of disease phenotypes in chronic lymphocytic leukemia. Nat Genet. 2016;48(3):253–264.
- Kulis M, Merkel A, Heath S, et al. Whole-genome fingerprint of the DNA methylome during human B cell differentiation. Nat Genet. 2015;47(7):746–756.
- Kulis M, Heath S, Bibikova M, et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat Genet. 2012;44(11):1236–1242.
- Kretzmer H, Biran A, Purroy N, et al. Preneoplastic alterations define CLL DNA methylome and persist through disease progression and therapy. Blood Cancer Discov. 2021;2(1):54–69.
- Mansouri L, Wierzbinska JA, Plass C, et al. Epigenetic deregulation in chronic lymphocytic leukemia: clinical and biological impact. Semin Cancer Biol. 2018;51:1–11.
- Queirós AC, Villamor N, Clot G, et al. A B-cell epigenetic signature defines three biologic subgroups of chronic lymphocytic leukemia with clinical impact. Leukemia. 2015;29(3):598–605.
- Wojdacz TK, Amarasinghe HE, Kadalayil L, et al. Clinical significance of DNA methylation in chronic lymphocytic leukemia patients: results from 3 UK clinical trials. Blood Adv. 2019;3(16):2474–2481.
- Damle RN, Wasil T, Fais F, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999;94(6):1840–1847.
- Hamblin TJ, Davis Z, Gardiner A, et al. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94(6):1848–1854.
- Hallek M, Cheson BD, Catovsky D, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131(25):2745–2760.
- Eichhorst B, Robak T, Montserrat E, et al. Chronic lymphocytic leukaemia: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2021;32(1):23–33.
- Hamblin TJ, Davis ZA, Oscier DG. Determination of how many immunoglobulin variable region heavy chain mutations are allowable in unmutated chronic lymphocytic leukaemia - long-term follow up of patients with different percentages of mutations. Br J Haematol. 2008;140(3):320–323.
- Janovska P, Poppova L, Plevova K, et al. Autocrine signaling by Wnt-5a deregulates chemotaxis of leukemic cells and predicts clinical outcome in chronic lymphocytic leukemia. Clin Cancer Res. 2016;22(2):459–469.
- Roman-Gomez J, Jimenez-Velasco A, Cordeu L, et al. WNT5A, a putative tumour suppressor of lymphoid malignancies, is inactivated by aberrant methylation in acute lymphoblastic leukaemia. Eur J Cancer. 2007;43(18):2736–2746.
- Ying J, Li H, Yu J, et al. WNT5A exhibits tumor-suppressive activity through antagonizing the Wnt/beta-catenin signaling, and is frequently methylated in colorectal cancer. Clin Cancer Res. 2008;14(1):55–61.
- Martín V, Valencia A, Agirre X, et al. Epigenetic regulation of the non-canonical Wnt pathway in acute myeloid leukemia. Cancer Sci. 2010;101(2):425–432.
- Li J, Ying J, Fan Y, et al. WNT5A antagonizes WNT/β-catenin signaling and is frequently silenced by promoter CpG methylation in esophageal squamous cell carcinoma. Cancer Biol Ther. 2010;10(6):617–624.
- Baskar S, Kwong KY, Hofer T, et al. Unique cell surface expression of receptor tyrosine kinase ROR1 in human B-cell chronic lymphocytic leukemia. Clin Cancer Res. 2008;14(2):396–404.
- Daneshmanesh AH, Mikaelsson E, Jeddi-Tehrani M, et al. Ror1, a cell surface receptor tyrosine kinase is expressed in chronic lymphocytic leukemia and may serve as a putative target for therapy. Int J Cancer. 2008;123(5):1190–1195.
- Fukuda T, Chen L, Endo T, et al. Antisera induced by infusions of autologous Ad-CD154-leukemia B cells identify ROR1 as an oncofetal antigen and receptor for Wnt5a. Proc Natl Acad Sci U S A. 2008;105(8):3047–3052.
- Kaucká M, Plevová K, Pavlová S, et al. The planar cell polarity pathway drives pathogenesis of chronic lymphocytic leukemia by the regulation of B-lymphocyte migration. Cancer Res. 2013;73(5):1491–1501.
- Hasan MK, Yu J, Chen L, et al. Wnt5a induces ROR1 to complex with HS1 to enhance migration of chronic lymphocytic leukemia cells. Leukemia. 2017;31(12):2615–2622.
- Hasan MK, Ghia EM, Rassenti LZ, et al. Wnt5a enhances proliferation of chronic lymphocytic leukemia and ERK1/2 phosphorylation via a ROR1/DOCK2-dependent mechanism. Leukemia. 2021;35(6):1621–1630.
- Yu J, Chen L, Cui B, et al. Wnt5a induces ROR1/ROR2 heterooligomerization to enhance leukemia chemotaxis and proliferation. J Clin Invest. 2016;126(2):585–598.
- Zhang Q, Wang HY, Liu X, et al. Cutting edge: ROR1/CD19 receptor complex promotes growth of mantle cell lymphoma cells independently of the B cell receptor-BTK signaling pathway. J Immunol. 2019;203(8):2043–2048.
- Cui B, Ghia EM, Chen L, et al. High-level ROR1 associates with accelerated disease progression in chronic lymphocytic leukemia. Blood. 2016;128(25):2931–2940.
- Bennett LB, Taylor KH, Arthur GL, et al. Epigenetic regulation of WNT signaling in chronic lymphocytic leukemia. Epigenomics. 2010;2(1):53–70.
- Rahmatpanah FB, Carstens S, Guo J, et al. Differential DNA methylation patterns of small B-cell lymphoma subclasses with different clinical behavior. Leukemia. 2006;20(10):1855–1862.
- Rahmatpanah FB, Carstens S, Hooshmand SI, et al. Large-scale analysis of DNA methylation in chronic lymphocytic leukemia. Epigenomics. 2009;1(1):39–61.
- Liu TH, Raval A, Chen SS, et al. CpG Island methylation and expression of the secreted frizzled-related protein gene family in chronic lymphocytic leukemia. Cancer Res. 2006;66(2):653–658.
- Chim CS, Fung TK, Wong KF, et al. Infrequent Wnt inhibitory factor-1 (Wif-1) methylation in chronic lymphocytic leukemia. Leuk Res. 2006;30(9):1135–1139.
- Maity PC, Bilal M, Koning MT, et al. is an inherited risk factor for CLL through the acquisition of a single-point mutation enabling autonomous BCR signaling. Proc Natl Acad Sci U S A. 2020;117(8):4320–4327.
- Minici C, Gounari M, Übelhart R, et al. Distinct homotypic B-cell receptor interactions shape the outcome of chronic lymphocytic leukaemia. Nat Commun. 2017;8:15746.
- Giacopelli B, Zhao Q, Ruppert AS, et al. Developmental subtypes assessed by DNA methylation-iPLEX forecast the natural history of chronic lymphocytic leukemia. Blood. 2019;134(8):688–698.
- Nadeu F, Royo R, Clot G, et al. IGLV3-21R110 identifies an aggressive biological subtype of chronic lymphocytic leukemia with intermediate epigenetics. Blood. 2021;137(21):2935–2946.