
ABSTRACT
Idiopathic pulmonary fibrosis (IPF) is a fatal interstitial lung disease with an unclear pathogenesis. This study aimed to elucidate the function and potential mechanisms of TUG1 in IPF progression. Cell viability and migration were detected by CCK-8 and transwell assays. Autophagy, fibrosis, or EMT-related proteins were measured by Western blotting. Pro-inflammatory cytokine levels were assessed by ELISA kits. The subcellular localization of TUG1 was observed by FISH assay. RIP assay detected the interaction between TUG1 and CDC27. TUG1 and CDC27 was up-regulated in TGF-β1-induced RLE-6TN cells. TUG1 depletion suppressed pulmonary fibrosis via attenuating inflammation, EMT, inducing autophagy and inactivating PI3K/Akt/mTOR pathway in vitro and in vivo. TUG1 knockdown prevented CDC27 expression. TUG1 silencing ameliorated pulmonary fibrosis by reducing CDC27 expression and inhibiting PI3K/Akt/mTOR pathway.
Introduction
Pulmonary fibrosis has been regarded as a common consequence of multiple interstitial lung diseases. Idiopathic pulmonary fibrosis (IPF), as a major kind of pulmonary fibrosis, is a life-threatening, chronic and fibrotic disorder with a median survival less than 4 years [Citation1]. As a progressive interstitial lung disease, IPF is featured by chronic non-specific interstitial inflammation and extensive deposition of collagen, which has been recognized to be a lethal and rapidly progressed fibrosis disease [Citation2]. It is currently accepted that IPF is attributed to the trans-differentiation of alveolar epithelial cells to myofibroblasts through epithelial-mesenchymal transition (EMT) and consequent extracellular matrix overproduction [Citation3], indicating the pivotal role of EMT in IPF. As estimated, nearly a third of fibroblasts originate from epithelial cells in pulmonary fibrosis [Citation4]. Recently, it was suggested that autophagy has been implicated in the pathological mechanisms of IPF [Citation5]. Enhancement of autophagy conferred inhibitory function in IPF [Citation6]. In addition, chronic inflammation can contribute to trans-differentiation to myofibroblasts and exacerbate IPF progression [Citation7]. Autophagy induction could protect against fibrosis via suppressing inflammation and EMT [Citation8]. Therefore, autophagy induction may provide a novel therapeutic strategy against IPF.
Long non-coding RNAs (lncRNAs), a class of non-coding RNAs longer than 200 nt, are critical regulators of various pathophysiological states [Citation9]. Recent reports suggested that lncRNAs took part in the pathogenesis of IPF [Citation10,Citation11]. LncRNA taurine upregulated gene 1 (TUG1) has been documented to contribute to fibrosis of many different organs, such as liver [Citation12], heart [Citation13], and kidney [Citation14]. However, whether lncRNA TUG1 can be implicated in IPF progression is still obscure. Gu et al. reported that lncRNA TUG1 was up-regulated in endometrial fibrosis, which conferred endometrial fibrosis and inflammation during the progression of intrauterine adhesion [Citation15]. Therefore, we speculated the potential oflncRNA TUG1 driving the development of IPF. So far, the regulation of TUG1 in IPF has not been elucidated.
Notably, bioinformatics analysis suggested an association between lncRNA TUG1 and CDC27. A recent study indicated that inhibition of CDC27 participated in hsa_circ_0044226 depletion-mediated protection against IPF [Citation16]. The interaction between lncRNA and protein has been widely documented. Lou et al. reported that lncRNA1 BS-DRL1 interacted with HMGB1 to maintain genome stability in neurons [Citation17]. A recent study revealed that lncRNA modulated endothelial permeability during diabetic retinopathy via interaction with PRKCB2 [Citation18]. But so far, the interaction between lncRNA TUG1 and CDC27 and its regulatory function in IPF have not been reported, which needs to be investigated.
PI3K/Akt/mTOR pathway has been widely accepted to be a key regulator of autophagy [Citation19], which may be an effective intervention for treating IPF. For example, suppression of autophagy could aggravate IL-17A-induced pulmonary fibrosis via inhibiting PI3K/Akt/mTOR pathway [Citation20]. Yu et al. showed that Yifei Sanjie formula treatment attenuated pulmonary fibrosis via inactivating PI3K/Akt/mTOR pathway to promote autophagy [Citation21]. More importantly, lncRNA TUG1 has been revealed to suppress the fibrosis of mesangial cells by inactivating PI3K/Akt pathway [Citation22]. Therefore, PI3K/Akt/mTOR signalling pathway might also be implicated in lncRNA TUG1-mediated regulation in IPF. So far, the regulation of lncRNA TUG1 in PI3K/Akt pathway has not been documented.
In this work, the functional roles of lncRNA TUG1 in IPF were investigated. We found that lncRNA TUG1 knockdown slowed down IPF progression via suppressing inflammation and EMT and enhancing autophagy by suppressing CDC27 expression and inactivating PI3K/Akt/mTOR pathway. These data identify lncRNA TUG1 as a promising target to impede IPF development.
Materials and methods
Cell culture and treatment
RLE-6TN cells were obtained from ATCC (Rockville, MD, USA), primary rat lung fibroblasts were purchased from Procell Life Science & Technology Co., Ltd. (Wuhan, China), which were maintained in DMEM with 10% foetal bovine serum (FBS, Hyclone, USA) at 37°C with 5% CO2. RLE-6TN cells and primary rat lung fibroblasts were incubated with TGF-β1 (10 ng/mL; PeproTech, Rocky Hill, NJ, USA) for 48 h before subsequent experiments. To inhibit autophagy, autophagy inhibitor 3-Methyladenine (3-MA) (1 mM, MCE, South Brunswick, NJ, USA) was adopted.
Cell transfection
ShRNAs targeting TUG1 (sh‐TUG1–1 and sh‐TUG1–2) and negative control shRNA (sh‐NC) were provided by Genepharma (Shanghai, China). For CDC27 overexpression, the plasmid containing the full‐length CDC27 cDNA was purchased from Sino Biological (Beijing, China). RLE-6TN cells were transfected with these segments using Lipofectamine 2000 (Thermo Fisher, Franklin Lakes, MA, USA).
CCK-8
RLE‐6TN cells with various treatments were seeded into 96‐well plates (2 × 10 [Citation4] cells per well). To determine cell proliferation, CCK-8 solution (MCE) was added to cells and incubated for 2 h. The OD value at 450 nm was acquired on a microplate reader (Hidex, Turku, Finland).
Fluorescent in situ hybridization (FISH)
The localization of TUG1 in RLE-6TN was determined by FISH. Briefly, RLE-6TN cells were seeded in a 24-well plate. After fixation and permeabilization, cells were probed with TUG1 probe. Following that, the cells were rinsed in a hybridization solution and stained with DAPI (Sigma-Aldrich, Saint Louis, MO, USA) in the dark. Images were obtained using a laser scanning confocal microscope (Leica, Germany).
Immunofluorescence staining
To detect LC3-II expression in RLE‐6TN cells, immunofluorescence staining was carried out. Cells fixed in 4% paraformaldehyde were immersed in 3% BSA for 1.5 h. Afterwards, incubation with LC3B antibody (ab48394, 1 µg/ml., Abcam) at 4°C overnight was performed. After probing with secondary antibody (bs-0369 M-FITC, 1:100, Bioss) for 1 h and nuclei staining with DAPI solution, the results were observed under a fluorescence microscope (Olympus, Tokyo, Japan).
Transwell assay
RLE‐6TN cells (2 × 104) received various treatments were added to the upper Transwell chamber without FBS. The lower chamber was added with DMEM containing 10% FBS. RLE‐6TN cells were incubated for 24 h for migration, and subjected to fixation in 4% paraformaldehyde followed by staining with 0.1% crystal violet (Solarbio, Beijing, China). The migratory cells were photographed under a light microscope (Olympus).
RNA immunoprecipitation (RIP)
To determine the interaction between lncRNA TUG1 and CDC27, RIP was carried out using the RIP kit (BersinBio, Guangzhou, China). In short, RLE‐6TN cells were lysed and added with magnetic beads conjugated with IgG or CDC27 antibody. After incubation overnight, the enrichment of lncRNA TUG1 in the bound RNA was assessed by qRT-PCR.
Animal model
Male Wistar rats (180–220 g) were obtained from Tianqin Biotechnology Co., Ltd. (Hunan, China) and randomly divided into control, bleomycin (BLM), BLM+ sh-NC, and BLM+ sh-TUG1 groups (n = 9 per group). The rats were intratracheally injected with 6 IU/kg BLM (Sigma Aldrich, St. Louis, MO, USA) [Citation23] after injection with pentobarbital sodium (50 mg/kg) to induce pulmonary fibrosis. The rats were administered with 30 μL of lentiviruses containing sh-NC or sh-TUG1 (5 × 10 [Citation8] TU, GeneChem, Shanghai, China) via intratracheal injection 1 day after BLM injection [Citation24]. The control rats were intratracheally injected with equal volume saline. On days 28 after BLM injection, all rats were sacrificed and lung tissues were acquired for further experiments. The experiment was approved by the Ethics Committee of Peking University Shenzhen Hospital.
Histological analysis
The collected lung tissues were embedded in paraffin and cut into 5-μm sections. Subsequently, routine haematoxylin-eosin (HE, Solarbio) and Masson’s trichrome staining (Solarbio) were performed to evaluate fibrotic changes in the tissues. For immunohistochemical analysis, the sections were incubated with primary antibodies α-SMA (ab124964, 1:2000, Abcam), collagen I (ab270993, 1:500, Abcam) overnight at 4°C. After that, the secondary antibody was applied. DAB substrate was used for visualization. The percentage of positive area was analysed using Image pro-plus (Media Cybernetics, Bethesda, MD, USA).
ELISA
TNF-α, IL-1β, and IL-6 levels in RLE‐6TN cells or lung tissues were measured using commercial ELISA kits (eBioscience, San Diego, CA, USA), following the manufacturer’s protocols.
Quantitative real-time PCR (qRT-PCR)
Total RNA from RLE‐6TN cells or lung tissues was acquired using Trizol (Thermo Fisher). Subsequently, PrimeScript reagent Kit (Takara, Tokyo, Japan) was adopted for cDNA synthesis. SYBR Premix EX Taq (TransGen, Beijing, China) was selected for qRT‐PCR. The gene levels normalized to β-actin were calculated using the 2−ΔΔCT method. The primer sequences are presented as follows:
LncRNA TUG 1: F:5’- CCAGACCCTCAGTGCAAACT-3’;
R:5’- TGCTTCTTGGGTCACAGTGG-3’;
CDC27:F: 5’- TGCTATATGGCAAGCACTAAACC-3’;
R:5’- CATTTTGCAAGCAGGTATTTACAC-3’;
β-actin:F: 5’- CGTTGACATCCGTAAAGACCTC-3’;
R:5’- TAGGAGCCAGGGCAGTAATCT-3’.
Western blotting
Proteins of lung tissues and RLE‐6TN cells were extracted with RIPA Lysis Buffer (Beyotime, Shanghai, China). After quantification, the protein samples were subjected to SDS-PAGE, then blotted onto polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA). The membranes were probed with the following primary antibodies LC3B (ab48394, 1 µg/ml, Abcam), Beclin-1 (ab207612, 1:2000, Abcam), p62 (ab109012, 1:10000, Abcam), E-cadherin (ab231303, 1 µg/ml, Abcam), N-cadherin (ab76011, 1:5000, Abcam), Vimentin (ab92547, 1:1000, Abcam), α-SMA (ab124964, 1:10000, Abcam), collagen I (ab270993, 1:1000, Abcam), Fibronectin 1 (ab45688, 1:1000, Abcam), p-PI3K (ab182651, 1:1000, Abcam), PI3K (ab191606, 1:1000, Abcam), p-Akt (ab38449, 1:500, Abcam), Akt (ab38449, 1:500, Abcam), p-mTOR (AP0094, 1:500, ABclonal), mTOR (A2445, 1:500, ABclonal), CDC27 (A3333, 1:1000, ABclonal), and GAPDH (ab8245, 1:2000, Abcam) at 4°C overnight, followed by incubation with secondary antibody (bs-0295 G-HRP, 1:1000, Bioss). ECL chemiluminescence (Beyotime) was applied for analysis.
Statistical analysis
All data from three independent experiments are presented as mean± standard deviation (SD). Student’s t test for two groups or One-Way ANOVA followed by Tukey post test for multiple groups was performed using SPSS 20.0 (IBM, NY, USA). P values less than 0.05 were considered statistically significant.
Results
Knockdown of lncRNA TUG1 promoted autophagy and inhibited inflammation in TGF-β1-challenged RLE-6TN cells
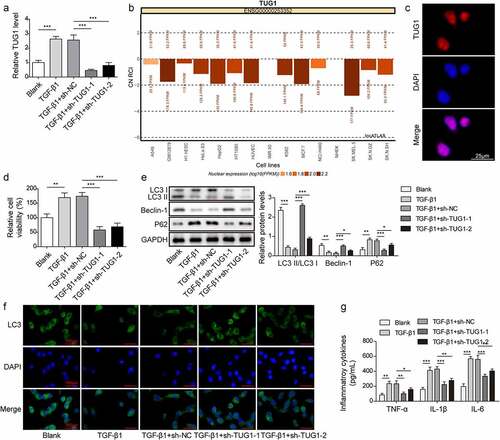
Although the biological functions of lncRNA TUG1 in fibrosis of various tissues, such as liver [Citation12], heart [Citation13], and endometrium [Citation15] have been verified, its modulation in IPF remains obscure. First, the level of lncRNA TUG1 in RLE-6TN cells after exposure to TGF-β1 was evaluated. qRT-PCR showed that lncRNA TUG1 was up-regulated in TGF-β1-induced RLE-6TN cells, which was remarkably restrained by sh-TUG1 (). It was predicted that TUG1 was mainly expressed in the cell nucleus using LncATLAS database (). Subsequently, RNA-FISH experiment further verified that the predominant nucleus localization of TUG1 in RLE-6TN cells (). Furthermore, RLE-6TN cell viability was enhanced after TGF-β1 induced, however, knockdown of TUG1 effectively suppressed the increased viability (). It has been shown that activation of autophagy by fluorofenidone attenuated paraquat‑induced pulmonary fibrosis [Citation25]. We found that TGF-β1 challenge deceased the LC3-II/I ratio and Beclin-1 level, but p62 level was increased, which was reversed by lncRNA TUG1 silencing (). Consistently, immunofluorescence staining indicated that lncRNA TUG1 depletion restored TGF-β1-mediated decreased of the expression of LC3 (). Besides, the increased levels of proinflammatory cytokines TNF-α, IL-1β, and IL-6 in TGF-β1-treated RLE-6TN cells were reduced by TUG1 down-regulation (). These findings suggested that TGF-β1-induced autophagy deficiency and inflammation in RLE-6TN cells were attenuated by lncRNA TUG1 silencing.
Figure 1. LncRNA TUG1 silencing promoted autophagy and inhibited inflammation in TGF-β1-exposed RLE-6TN cells.
Knockdown of lncRNA TUG1 suppressed EMT and fibrinogenesis in TGF-β1-stimulated RLE-6TN cells
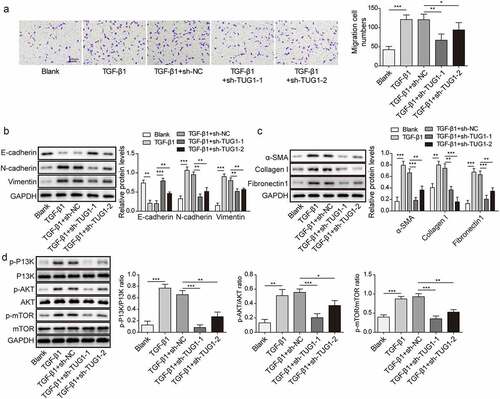
EMT has been confirmed to be implicated in the pathogenesis of pulmonary fibrosis [Citation26]. Therefore, we further explored the role of lncRNA TUG1 in TGF-β1-induced fibrinogenesis and EMT in RLE-6TN cells. The migration of RLE-6TN cells was promoted by TGF-β1, which could be repressed by sh-TUG1 (). Moreover, TGF-β1 exposure resulted in EMT in RLE-6TN cells via reducing E-cadherin expression, while enhancing N-cadherin and Vimentin expression, whereas TUG1 silencing abolished these changes in EMT markers (). In addition, myofibroblast markers α-SMA, collagen I and Fibronectin 1 levels were assessed by Western blotting to evaluate pulmonary fibrosis. As depicted in , depletion of lncRNA TUG1 significantly reversed the elevated α-SMA, collagen I and Fibronectin 1 levels in TGF-β1-exposed RLE-6TN cells (). Alveolar epithelial cells can regenerate the alveolar architecture to transdifferentiate into activated fibroblasts through EMT process [Citation27]. Thus, primary rat lung fibroblasts were adopted to evaluate the role of TUG1 in pulmonary fibrosis. TGF-β1 exposure strikingly increased TUG1 expression level, while TUG1 knockdown abolished this change (Fig. S1A). Moreover, lung fibroblast viability and levels of myofibroblast markers (α-SMA, collagen I and Fibronectin 1) were enhanced by TGF-β1 stimulation, which were abrogated after TUG1 knockdown (Fig. S1B-S1C). Results suggested that TUG1 could exert antifibrotic effects in fibroblasts. Moreover, the aberrant PI3K/Akt mTOR signalling activation has been proved in pulmonary fibrosis [Citation28]. TGF-β1 led to an enhancement in PI3K/Akt mTOR pathway activation, which was suppressed by sh-TUG1 (). The above results revealed that lncRNA TUG1 silencing repressed TGF-β1-induced fibrinogenesis, EMT, and PI3K/Akt/mTOR pathway activation in RLE-6TN cells.
Figure 2. LncRNA TUG1 depletion suppressed EMT and fibrinogenesis in TGF-β1-stimulated RLE-6TN cells.
3-MA counteracted the inhibition of TUG1 knockdown in TGF-β1-induced inflammation, EMT and pulmonary fibrosis
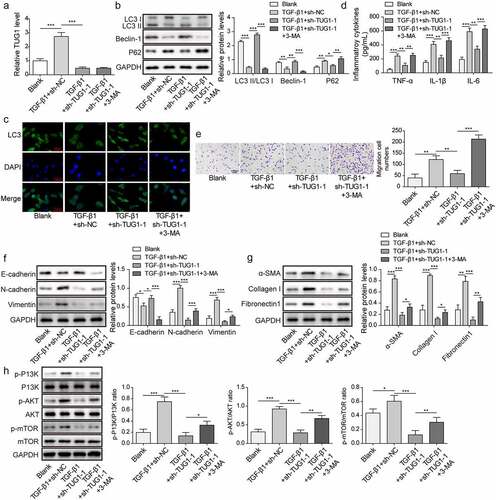
To elucidate the involvement of autophagy in the anti-fibrotic efficacy of TUG1 depletion, 3-MA, an inhibitor of autophagy was applied. After lncRNA TUG1 knockdown, 3-MA administration did not affect TUG1 expression in TGF-β1-exposed RLE-6TN cells (). As expected, the autophagy-promoting effect of sh-TUG1 on TGF-β1-treated RLE-6TN cells was counteracted by 3-MA as confirmed by reducing LC3-II/I ratio, Beclin-1 and LC3 expression, and enhancing p62 expression ()). In addition, 3-MA treatment obviously elevated TNF-α, IL-1β, and IL-6 levels in TUG1-silenced RLE-6TN cells, indicating that the anti-inflammatory effects of TUG1 knockdown was reversed by 3-MA (). Similarly, the inhibition in migration by sh-TUG1 was attenuated by 3-MA in RLE-6TN cells exposed to TGF-β1 (). Furthermore, 3-MA facilitated EMT of TUG1-depleted RLE-6TN cells (). Accordingly, the suppression of sh-TUG1 in expression of fibrotic markers α-SMA, fibronectin 1 and collagen I was evidently abolished by 3-MA (). LncRNA TUG1 silencing-induced dephosphorylation of PI3K, Akt, and mTOR was repressed after administration with 3-MA (). These observations indicated that TUG1 depletion protected against TGF-β1-induced inflammation, EMT, and pulmonary fibrosis via inducing autophagy.
Figure 3. 3-MA abolished the protection of TUG1 knockdown against TGF-β1-induced inflammation, EMT and fibrosis in RLE-6TN cells.
LncRNA TUG1 interacted with CDC27 in TGF-β1-stimulated RLE-6TN cells
Next, the potential mechanisms by which TUG1 knockout relieved fibrosis was investigated. RPISeq database predicted that TUG1 exhibited a high affinity for CDC27 (RF classifier: 0.85 score; SVM classifier: 0.98 score). RIP assay further indicated that TUG1 was highly enriched in CDC27 antibody group as compared with IgG (). More importantly, CDC27 levels were strikingly lowered after TUG1 depletion in RLE-6TN cells (). Therefore, lncRNA TUG1 could play pivotal roles in TGF-β1-challenged RLE-6TN cells by directly interacting with CDC27.
Figure 4. LncRNA TUG1 interacted with CDC27 in TGF-β1-exposed RLE-6TN cells.

CDC27 overexpression reversed the anti-fibrotic effect of lncRNA TUG1 silencing on TGF-β1-induced RLE-6TN cells
To investigate whether CDC27 was involved in the effect of lncRNA TUG1 in TGF-β1-stimulated RLE-6TN cells, we upregulated the expression of CDC27 and performed rescue experiments: sh-TUG1+Oe-NC group and sh-TUG1+Oe-CDC27 group. As presented in , overexpression of CDC27 remarkably up-regulated CDC27 level in TUG1-silenced RLE-6TN cells; however, TUG1 expression was not affected by enforced expression of CDC27. CDC27 overexpression enhanced the viability of RLE-6TN cell exposure to TGF-β1, which also reversed sh-TUG1-mediated weakened viability (). In addition, CDC27 overexpression led to autophagy inhibition in TGF-β1-exposed cells, and silencing of TUG1-induced autophagy promotion was weakened by CDC27 overexpression (. Besides, TGF-β1-induced inflammation and migration was further promoted by CDC27 overexpression, which also reversed the inhibitory effects of sh-TUG1 on inflammation and migration (). Consistently, overexpression of CDC27 further facilitated EMT, expression of fibrotic markers, and PI3K/Akt/mTOR pathway activation in RLE-6TN cells stimulated with TGF-β1, which also reversed the inhibition effect of lncRNA TUG1 knockdown in EMT, pulmonary fibrosis, and PI3K/Akt/mTOR pathway activation ()). Taken together, CDC27 was involved in lncRNA TUG1 depletion-mediated anti-fibrotic function in RLE-6TN cells.
Figure 5. CDC27 overexpression reversed the effect of lncRNA TUG1 silencing on autophagy and inflammation in TGF-β1-stimulated RLE-6TN cells.
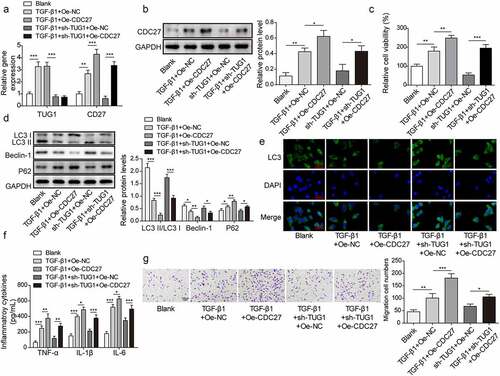
Figure 6. Overexpression of CDC27 counteracted lncRNA TUG1 depletion-mediated inhibition in EMT and fibrinogenesis in TGF-β1-exposed RLE-6TN cells.
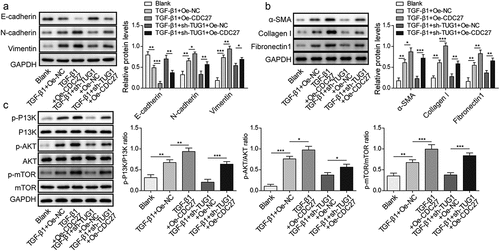
Knockdown of lncRNA TUG1 improved BLM-induced IPF in rats
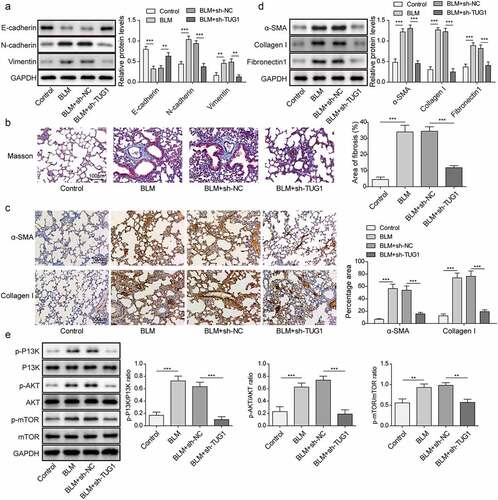
Having demonstrated that lncRNA TUG1 deficiency could protect against fibrinogenesis in vitro, we further sought to evaluate the beneficial effect of lncRNA TUG1 silencing on pulmonary fibrosis in vivo. BLM was used to induce pulmonary fibrosis in rats for this purpose. LncRNA TUG1 level was strikingly elevated by BLM in lung tissues, whereas this change could be reversed by sh-TUG1 (). As assessed by HE staining, BLM resulted in lobular interstitium thickness and inflammatory cell infiltration in rats, which was alleviated by lncRNA TUG1 deficiency (). The lungs of rats after treatment BLM presented declined LC3-II/I ratio and Beclin-1 expression, but enhanced p62 expression, however, sh-TUG1 relieved BLM-induced autophagy deficiency (). Likewise, knockdown of TUG1 counteracted BLM-mediated increase in TNF-α, IL-1β, and IL-6 levels (). Moreover, CDC27 level was up-regulated by BLM treatment in lung tissues, which was reduced after TUG1 knockdown (). In addition, BLM resulted in EMT in lung tissues of rats, which was reversed by sh-TUG1 (). Masson’s trichrome staining showed that the fibrotic lesions were developed in BLM group, and in contrast, these fibrotic lesions were significantly ameliorated by depletion of TUG1 (). Accordingly, α-SMA, fibronectin 1, and collagen I expressions were reduced in lncRNA TUG1-silenced rats (). Furthermore, the enhancement in phosphorylation of PI3K, Akt, and mTOR after exposure to BLM was reversed by sh-TUG1 (). Collectively, our results indicated that depletion of TUG1 ameliorated pulmonary fibrosis in rats via activating autophagy, restraining inflammation and EMT, which were in line with the in vitro experiments.
Figure 7. Knockdown of lncRNA TUG1 restrained BLM-induced autophagy inhibition and inflammation in rats.
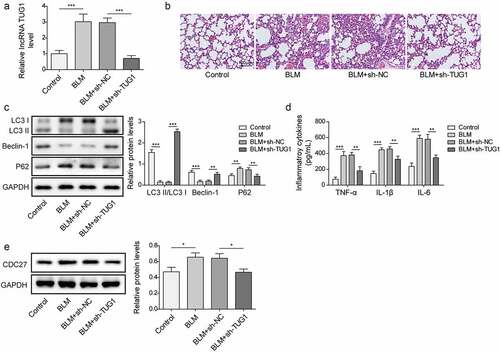
Figure 8. LncRNA TUG1 silencing suppressed EMT and pulmonary fibrosis in BLM-induced IPF in rats.
Discussion
IPF is a fatal lung disease, whose pathological mechanisms are poorly understood. The current treatments for reversing or halting the progression of IPF remain limited. Therefore, exploring effective antifibrotic therapy especially for impeding early-stage pulmonary fibrosis is highly needed. Here, we showed that TUG1 was up-regulated in the in vitro and in vivo models of IPF. LncRNA TUG1 down-regulation mitigated pulmonary fibrosis via repressing inflammatory response, EMT and activating autophagy through inhibiting CDC27 expression and subsequent inactivation of PI3K/Akt/mTOR pathway. Therefore, targeting lncRNA TUG1 may be a promising therapy for IPF.
Autophagy deficiency has been verified to contribute to multiple human diseases, including IPF [Citation29]. IPF has been considered as an ageing-related disorder caused by decreased autophagic activity and its consequence for accumulating impaired macromolecules and organelles [Citation30]. Consistent with these observations, we found autophagy inhibition in in vitro and in vivo models, whereas TUG1 knockdown effectively ameliorated autophagy deficiency. As previously reported, decreased TUG1 expression conferred LPS-induced autophagy in vascular endothelial cells [Citation31]. Lv et al. demonstrated that TUG1 depletion could facilitate autophagy in renal cell carcinoma cells [Citation32]. These studies further supported our results that deficiency of autophagy due to TUG1 overexpression facilitated IPF progression. mTOR is a major autophagy regulator, and its phosphorylation can restrain autophagy-related protein expression [Citation33]. Inactivation of PI3K/Akt pathway and its downstream mTOR could increase LC3-II and Beclin-1 expressing, suggesting the induction of autophagy [Citation34]. Consistent with these studies, we demonstrated that TUG1 depletion repressed TGF-β or BLM-induced PI3K/Akt/mTOR activation in vitro and in vivo. Thus, TUG1 depletion induced autophagy via inactivating PI3K/Akt/mTOR pathway, which slowed down IPF development.
Growing evidence has suggested that IPF has close relationship with inflammation [Citation35]. Particularly, the release of IL-1β drives fibrosis progression via promoting the production of IL-6 and TNF-α, which results in alveolar architecture disruption via enhancing fibroblast proliferation and collagen deposition [Citation36]. Given the contribution of inflammation to IPF progression, we first explored the regulation of TUG1 in inflammation during IPF. Our observations indicated that TUG1 silencing repressed the expression of IL-1β, IL-6, and TNF-α. It has been proved that EMT is responsible for the activation of fibroblasts and subsequent trans-differentiation into myofibroblasts in IPF [Citation37]. EMT is characterized by reducing epithelial markers, such as E-cadherin and enhancing mesenchymal markers including vimentin and N-cadherin [Citation38]. In this study, E-cadherin expression was raised and vimentin and N-cadherin expression was reduced by silencing of TUG1 in response to TGF-β or BLM in vitro and in vivo. Of note, the interplay between autophagy and inflammation or EMT has been documented. Hill et al. showed that autophagy deficiency could induce EMT, which drove the development of IPF [Citation39]. Induction of autophagy by oestrogen receptor alpha could inhibit atherosclerosis via suppressing inflammation [Citation40]. It has been reported that alveolar epithelial cells can regenerate the alveolar architecture to transdifferentiate into activated fibroblasts through EMT process [Citation27]. Fibroblast activation exerts a key role in the development of pulmonary fibrosis [Citation41]. Therefore, we further explored the effect of TUG1 on the activation of primary rat lung fibroblasts. Our results revealed that TUG1 deficiency repressed TGF-β-induced proliferation and activation of rat lung fibroblasts. Furthermore, inhibition of autophagy could reverse TUG1 silencing-mediated protection against fibrosis via enhancing EMT and inflammation in RLE-6TN cells. Therefore, TUG1 promoted IPF via deficient autophagy-mediated EMT and inflammation.
To further elucidate the mechanisms underlying TUG1-mediated autophagy inhibition, inflammation, and EMT during IPF progression, CDC27 was focused on. LncRNAs can exert biological functions via the transcriptional modulation of gene expression. For example, lncRNA SNHG11 was found to bind to HIF-1α target gene promoter and promote transcription during colorectal cancer invasion and metastasis [Citation42]. Zhao et al. suggested that lncRNA TMPO-AS1 bond to E2F6 and facilitated LCN2 transcription, which contributed to ovarian cancer development [Citation43]. In this study, the binding between TUG1 and CDC27 was validated by RIP assay. Moreover, TUG1 knockdown down-regulated the mRNA and protein level of CDC27. Given that CDC27 inhibition was documented to attenuate pulmonary fibrosis via suppressing EMT [Citation16], we hypothesized that TUG1 might accelerate IPF via promoting CDC27 transcription and expression. In line with the hypothesis, our findings indicated that CDC27 overexpression abolished the protection of TUG1 knockdown against fibrosis via exacerbating autophagy inhibition, inflammation, and EMT.
In conclusion, our findings demonstrated that knockdown of lncRNA TUG1 alleviated pulmonary fibrosis progression via promoting autophagy and suppressing inflammation and EMT by inhibiting CDC27 expression and inactivating PI3K/AKT/mTOR pathway. Our observations shed light on the pathogenesis of IPF and identify lncRNA TUG1 as a potential therapeutic target for IPF.
Abbreviations
| IPF | = | idiopathic pulmonary fibrosis |
| EMT | = | epithelial-mesenchymal transition |
| lncRNAs | = | long non-coding RNAs |
| TUG1 | = | taurine upregulated gene 1 |
| 3-MA | = | 3-Methyladenine |
| sh‐NC | = | negative control shRNA |
| sh‐TUG1 | = | shRNA targeting TUG1 |
| FISH | = | Fluorescent in situ hybridization |
| RIP | = | RNA immunoprecipitation |
| BLM | = | bleomycin |
| SD | = | standard deviation |
| HE | = | haematoxylin-eosin. |
Authors’ contribution
FQ: Conceptualization; Writing-original draft; Methodology; Formal analysis;
ZDL: Supervision; Validation;
WDH: Data curation; Resources;
SCW: Investigation; Software;
XML: Visualization; Project administration;
WDS: Funding acquisition; Writing-review & editing.
All authors have read and approved the final version of this manuscript to be published.
Availability of Data and Material
All data generated or analysed during this study are included in this article. The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request
Consent for publication
Not Applicable. This article does not contain any studies with human participants performed by any of the authors.
Ethical approval
The experiment was approved by the Ethics Committee of Peking University Shenzhen Hospital.
Supplemental Material
Download Zip (567.9 KB)Acknowledgments
We would like to give our sincere gratitude to the reviewers for their constructive comments.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15592294.2023.2195305.
Additional information
Funding
References
- Ikeda K, Chiba H, Nishikiori H, et al. Serum surfactant protein D as a predictive biomarker for the efficacy of pirfenidone in patients with idiopathic pulmonary fibrosis: a post-hoc analysis of the phase 3 trial in Japan. Respir Res. 2020;21:316.
- Antoniu SA. Targeting the endothelin pathway in the idiopathic pulmonary fibrosis: the role of bosentan. Expert Opin Ther Targets. 2008;12:1077–15.
- Qian W, Cai X, Qian Q, et al. Angelica sinensis polysaccharide suppresses epithelial-mesenchymal transition and pulmonary fibrosis via a DANCR/AUF-1/FOXO3 regulatory axis. Aging Dis. 2020;11:17–30.
- Diaz-Pina G, Ordonez-Razo RM, Montes E, et al. The role of ADAR1 and ADAR2 in the regulation of miRNA-21 in idiopathic pulmonary fibrosis. Lung. 2018;196:393–400.
- Lv X, Li K, Hu Z. Autophagy and pulmonary fibrosis. Adv Exp Med Biol. 2020;1207:569–579.
- Kim MS, Baek AR, Lee JH, et al. IL-37 attenuates lung fibrosis by inducing autophagy and regulating TGF-beta1 production in mice. J Immunol. 2019;203:2265–2275.
- Araki K, Kinoshita R, Tomonobu N, et al. The heterodimer S100A8/A9 is a potent therapeutic target for idiopathic pulmonary fibrosis. J Mol Med (Berl). 2021;99:131–145.
- Wang YJ, Chen YY, Hsiao CM, et al. Induction of autophagy by pterostilbene contributes to the prevention of renal fibrosis via attenuating NLRP3 inflammasome activation and epithelial-mesenchymal transition. Front Cell Dev Biol. 2020;8:436.
- Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–874.
- Wang F, Li P, Li FS. Integrated analysis of a gene correlation network identifies critical regulation of fibrosis by lncRnas and TFs in idiopathic pulmonary fibrosis. BioMed Res Int. 2020;2020:6537462.
- Hadjicharalambous MR, Lindsay MA. Idiopathic pulmonary fibrosis: pathogenesis and the emerging role of long non-coding RNAs. Int J Mol Sci. 2020;21:524.
- Han X, Hong Y, Zhang K. TUG1 is involved in liver fibrosis and activation of HSCs by regulating miR-29b. Biochem Biophys Res Commun. 2018;503:1394–1400.
- Zhu Y, Feng Z, Jian Z, et al. Long noncoding RNA TUG1 promotes cardiac fibroblast transformation to myofibroblasts via miR29c in chronic hypoxia. Mol Med Rep. 2018;18:3451–3460.
- Wang F, Gao X, Zhang R, et al. LncRNA TUG1 ameliorates diabetic nephropathy by inhibiting miR-21 to promote TIMP3-expression. Int J Clin Exp Pathol. 2019;12:717–729.
- Ai Y, Chen M, Liu J, et al. lncRNA TUG1 promotes endometrial fibrosis and inflammation by sponging miR-590-5p to regulate Fasl in intrauterine adhesions. Int Immunopharmacol. 2020;86:106703.
- Qi F, Li Y, Yang X, et al. Hsa_circ_0044226 knockdown attenuates progression of pulmonary fibrosis by inhibiting CDC27. Aging (Albany NY). 2020;12:14808–14818.
- Lou MM, Tang XQ, Wang GM, et al. Long noncoding RNA BS-DRL1 modulates the DNA damage response and genome stability by interacting with HMGB1 in neurons. Nat Commun. 2021;12:4075.
- Sehgal P, Mathew S, Sivadas A, et al. LncRNA VEAL2 regulates PRKCB2 to modulate endothelial permeability in diabetic retinopathy. Embo J. 2021;40:e107134.
- Xu Z, Han X, Ou D, et al. Targeting PI3K/AKT/mTOR-mediated autophagy for tumor therapy. Appl Microbiol Biotechnol. 2020;104:575–587.
- Cong LH, Li T, Wang H, et al. IL-17A-producing T cells exacerbate fine particulate matter-induced lung inflammation and fibrosis by inhibiting PI3K/Akt/mTOR-mediated autophagy. J Cell Mol Med. 2020;24:8532–8544.
- Yu JZ, Ying Y, Liu Y, et al. Antifibrotic action of Yifei Sanjie formula enhanced autophagy via PI3K-AKT-mTOR signaling pathway in mouse model of pulmonary fibrosis. Biomed Pharmacother. 2019;118:109293.
- Zang XJ, Li L, Du X, et al. LncRNA TUG1 inhibits the proliferation and fibrosis of mesangial cells in diabetic nephropathy via inhibiting the PI3K/AKT pathway. Eur Rev Med Pharmacol Sci. 2019;23:7519–7525.
- Ramezani S, Javadi I, Kokhdan EP, et al. Protective and therapeutic effects of ethanolic extract of Nasturtium officinale (watercress) and vitamin E against bleomycin-induced pulmonary fibrosis in rats. Res Pharm Sci. 2021;16:94–102.
- Wang X, Chen Y, Lv L, et al. Silencing CD36 gene expression results in the inhibition of latent-TGF-beta1 activation and suppression of silica-induced lung fibrosis in the rat. Respir Res. 2009;10:36. DOI:10.1186/1465-9921-10-36.
- Jiang F, Li S, Jiang Y, et al. Fluorofenidone attenuates paraquatinduced pulmonary fibrosis by regulating the PI3K/Akt/mTOR signaling pathway and autophagy. Mol Med Rep. 2021;23:405. DOI:10.3892/mmr.2021.12044.
- Wei J, Zhao Q, Yang G, et al. Mesenchymal stem cells ameliorate silica-induced pulmonary fibrosis by inhibition of inflammation and epithelial-mesenchymal transition. J Cell Mol Med. 2021;25:6417–6428.
- Harada T, Nabeshima K, Hamasaki M, et al. Epithelial-mesenchymal transition in human lungs with usual interstitial pneumonia: quantitative immunohistochemistry. Pathol Int. 2010;60:14–21.
- Wanas H, El Shereef Z, Rashed L, et al. Ticagrelor ameliorates bleomycin-induced pulmonary fibrosis in rats bythe inhibition of TGF-β1/Smad3 and PI3K/AKT/mTOR pathways. Curr Mol Pharmacol. 2022;15:227–238.
- Romero Y, Bueno M, Ramirez R, et al. mTORC1 activation decreases autophagy in aging and idiopathic pulmonary fibrosis and contributes to apoptosis resistance in IPF fibroblasts. Aging Cell. 2016;15:1103–1112.
- Lopez-Otin C, Blasco MA, Partridge L, et al. The hallmarks of aging. Cell. 2013;153:1194–1217.
- Dong Y, Fan G, Li Y, et al. TUG1 represses apoptosis, autophagy, and inflammatory response by regulating miR-27a-3p/slit2 in LPS-Treated vascular endothelial cells. J Surg Res. 2020;256:345–354.
- Lv D, Xiang Y, Yang Q, et al. Long non-coding RNA TUG1 promotes cell proliferation and inhibits cell apoptosis, autophagy in clear cell renal cell carcinoma via MiR-31-5p/flot1 axis. Onco Targets Ther. 2020;13:5857–5868.
- Yu L, McPhee CK, Zheng L, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465:942–946.
- Wang L, Yang J, Wang HN, et al. LncRNA BCYRN1-induced autophagy enhances asparaginase resistance in extranodal NK/T-cell lymphoma. Theranostics. 2021;11:925–940.
- Borthwick LA. The IL-1 cytokine family and its role in inflammation and fibrosis in the lung. Semin Immunopathol. 2016;38:517–534.
- Kolb M, Margetts PJ, Anthony DC, et al. Transient expression of IL-1beta induces acute lung injury and chronic repair leading to pulmonary fibrosis. J Clin Invest. 2001;107:1529–1536.
- Yamada A, Aki T, Unuma K, et al. Paraquat induces epithelial-mesenchymal transition-like cellular response resulting in fibrogenesis and the prevention of apoptosis in human pulmonary epithelial cells. PLoS ONE. 2015;10:e0120192.
- Zhou W, Mo X, Cui W, et al. Nrf2 inhibits epithelial-mesenchymal transition by suppressing snail expression during pulmonary fibrosis. Sci Rep. 2016;6:38646.
- Hill C, Li J, Liu D, et al. Autophagy inhibition-mediated epithelial-mesenchymal transition augments local myofibroblast differentiation in pulmonary fibrosis. Cell Death Dis. 2019;10:591.
- Meng Q, Li Y, Ji T, et al. Estrogen prevent atherosclerosis by attenuating endothelial cell pyroptosis via activation of estrogen receptor alpha-mediated autophagy. J Adv Res. 2021;28:149–164.
- Geng Y, Li L, Yan J, et al. PEAR1 regulates expansion of activated fibroblasts and deposition of extracellular matrix in pulmonary fibrosis. Nat Commun. 2022;13:7114.
- Xu L, Huan L, Guo T, et al. LncRNA SNHG11 facilitates tumor metastasis by interacting with and stabilizing HIF-1alpha. Oncogene. 2020;39:7005–7018.
- Zhao H, Ding F, Zheng G. LncRNA TMPO-AS1 promotes LCN2 transcriptional activity and exerts oncogenic functions in ovarian cancer. Faseb J. 2020;34:11382–11394.