
Abstract
Novel phenylamine-substituted derivatives, possessing different photochromic groups, were investigated. The electrochemical activity of the monomers and polymers were studied by usage of the electrochemical and spectroelectrochemical measurements. Heterocyclic side rings influence on the oxidation potential of monomers, decreasing its value below oxidation potential in comparison to parent phenylamine units. The removal of electron is easier for diphenylamine derivatives than for the triphenylamine ones. Electroactivity of monomers allows to polymerize them into macromolecule chains. Triphenylamine-based polymers are more stable as a result of the enlarged conjugated systems. Furthermore, the energy gap value of the final polymer can be reduced by introduction of additional branch into the monomer structure.
Introduction
Electrochemically active conducting polymers can be deposited in electrochemical polymerization of heterocyclic or benzoid monomers [Citation1] mostly thiophene, pyrrole, furane or aniline. Due to its good electrochemical response,[Citation2] electrochromic properties [Citation3] and good environmental stability [Citation4] poly(aniline) (PANI) is known as a technologically important material in sensors, electrochromic display devices or corrosion protecting films. Many benzene ring or N-substituted derivatives were synthesized, e.g. poly(diphenylamine) (p(DPN)) [Citation5,6] to improve the processability,[Citation7] electrochromism [Citation8] or ion-exchange capability.[Citation9] For p(DPN), some additional type of redox transitions have been shown in comparison to PANI. After first stage of DPN oxidation, the presence of diphenylbenzidine cation radical (DPN⋅+) and after further oxidation, diphenoquinone diimine unit (DPN++) was revealed.[Citation5–11] Further modification of the structure of monomer unit led to substitution of the central nitrogen atom with third phenyl ring. The triphenylamine derivatives have been under study since the beginning of the 90s when the linear and starburst oligomers were synthesized and applied in the single-layer LEDs.[Citation12] Simple, branched or star-shape triarylamines were reported to act as effective hole-transporting materials.[Citation13–15] They were however usually either nonluminescent or barely emitting as a result of the inherent reductive quenching. Attachment of different heterocyclic rings next to arylamine unit was supposed to improve this emission characteristic.[Citation16]
Conjugated three-dimensional systems like poly(triphenylamine) (p(TPN)) have been the subject of studies mainly due to their excellent hole transport capacity.[Citation17,18] There are multiple pathways for the carrier to provide the electronic conductivity in the structure of this polymer. Consequently, the large amount of polymers with significantly better redox activity was formed.[Citation19–21] Moreover, promising properties like high stability during repetitive oxidation–reduction cycles [Citation22] have been shown by electrically active film of polytriphenylamine.
In the current study, we present electrochemical and spectroelectrochemical survey on the triphenylamine and diphenylamine derivatives (structures shown in Figure ) and their corresponding polymers. The aim of the study is to investigate the influence of the side rings on the electropolymerization process.
Figure 1. Structures of investigated monomers.
Materials and methods
Reagents and materials
The electrolyte solution was prepared by dissolution of tetrabutylammonium tetrafluoroborate Bu4NBF4 salt (TBABF4) (Janssen Chimica, 99.0%) in acetonitryle (ACN) (ACN for DNA synthesis and peptides, water <10 ppm, POCh, Gliwice, Poland). Concentration of the solution was 0.1 M and the measurements were carried out at room temperature.
Concentration of the compound was 0.5 mM and the measurements were done in three-electrode cell while using 2.0 mL sample solution. Cyclic voltammetry (CV) of electrodeposited film was taken in monomer-free solutions of the same supporting electrolyte. The solutions were carefully purged with argon bubbling for 30.0 min prior to the measurements.
The investigated monomers are N,N,N-tri(4-thienylphenyl)amine (TTN), N,N,N-tri(4-furanylphenyl)amine (TFN), N,N,N-tri(4-pyridylphenyl)amine (TPN), N,N,N-tri[4-(3,4-ethylendioxythienyl)phenyl]amine (TEN), N,N-bis(4-thienylphenyl)-N-butylamine (BTFA), N,N-bis(4-furanylphenyl)-N-butylamine (BFFA), N,N-bis(4-pyridylphenyl)-N-butylamine (BPFA), N,N-bis[4-(3,4-ethylendioxythienyl)phenyl]-N-butylamine (BEFN). For clarity, within the scope of the article the abbreviation of chemical names are used. The details of synthetic route for the monomers were previously described.[Citation23,24]
Instrumentation
Electrosynthesis and studies of polymers were performed on Ecochemie AUTOLAB potentiostat – galvanostat model PGSTAT20 driven by a computer. CV was used as an measurement method. Results were analysed using GPES programme (General Purpose Electrochemical System). Polymer films were synthesized directly on the Pt (the surface area = 13.0 mm2) used as a working electrode. Ag wire served as a pseudoreference electrode and platinum spiral was employed as an auxiliary electrode. Potentials were referenced to the redox potential of the ferrocene couple (Fc/Fc+) as an internal standard.
Spectroelectrochemical measurements were carried out by using UV–vis spectrophotometer Cintra 5 connected with OMNI 9, Cypress 2000 potentiostat. Data were analysed using OriginPro 7.5 programme. ITO-covered glass electrode served as a working electrode.
An atomic force microscope (Hysitron, TI-950 Triboindenter equipped with an AFM Q-Scope 250 Quesant Instruments) was used to measure the surface morphology of films. Tapping-mode AFM images of ultrathin films were recorded and the dates were analysed with SPM programme.
Results and discussion
Electrochemical activity of monomers
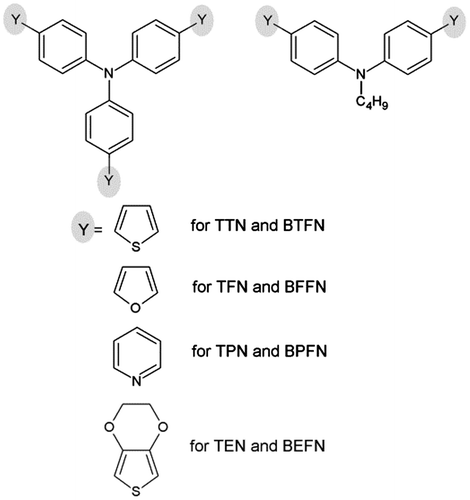
Multiple oxidation processes were exhibited by the CV curves of the investigated monomers. They were measured in ACN and the TBABF4 was used as the supporting electrolyte. Figures and depict characteristic cyclic voltammograms recorded during sweeping monomers solutions within either wide (Figure ) or narrow (Figure ) potential range. The first oxidation peak located within the potential range (0.30–0.50) V is almost irreversible. However, for triphenylamine derivatives, it seems to be less irreversible than for diphenylamine ones. The reason for this is the additional phenyl ring as it introduces more steric hindrance, which is known for native triphenylamine molecule.[Citation25] Additionally voltammograms shown in Figure (a) reveal reverse peaks shifted negative in comparison to the oxidation wave. That indicates an ECE mechanism for the electrode reaction, where after electrochemical oxidation, subsequent chemical reaction takes place. The products of this reaction are electrochemically active and also undergo electrochemical oxidation.
Figure 2. CV of (a) diphenylamines and (b) triphenylamines in 0.1 M solution of TBABF4 in ACN (ACN = acetonitrile) in wide potential range. Potential sweep rate v = 0.050 V s−1.
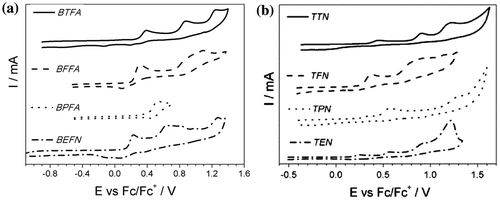
Figure 3. CV of (a) diphenylamines and (b) triphenylamines in 0.1 M solution of TBABF4 in ACN in narrow potential range. Potential sweep rate v = 0.050 V s−1.
The first oxidation peak recorded for the monomer’s solution is located in the range (0.233–0.534) V and (0.258–0.574) V for diphenylamines and triphenylamines, respectively. It illustrates the removal of electrons from the conjugated hetrocyclic-phenylamine backbone, resulting in radical cation formation. Monomeric arylamines are widely known to form stable radical cations upon chemical or electrochemical oxidation.[Citation24–40] Oxidation potential of diphenylamine unit to corresponding radical cation is Eox = 0.70 V (vs. Pt/(I−, ), 0.10 M TBAPF6, ACN) [Citation41] or 0.55 V (vs. Fc/Fc+, 0.1 M TBAPF6, ACN) [Citation42] and for triphenylamine Eox = 0.54 V (vs. Fc/Fc+ in DCM),[Citation43] 0.47 V (vs. Fc/Fc+, ACN),[Citation44,45] 0.53 V (vs. Fc/Fc+ in DCM/nBu4NPF6 (0.1 M)).[Citation46] It is known that for triphenylamine radical cation, the unpaired electron is localized mainly on the central amine nitrogen atom of TPA.[Citation25,47] However, the shift in oxidation potentials for investigated molecules confirms that the conjugated systems are delocalized within the molecule structure. It also overlaps side heterocyclic rings.
The effects of the heterocyclic side rings of the phenylamine core on the oxidation potential were examined and the data are gathered in Table . The presence of 3,4-ethylenedioxthiophene (EDOT) side rings reduces this potential, while furan and thiophene derivatives require application of higher potential range. The potential difference between the substituted phenylamine derivatives and the unsubstituted ones can be interpreted in terms of the electrophilicity changes. Electrophilicity can be induced by the electron-donating substituents or the electron-withdrawing ones to the lone pair of nitrogen atom of the phenylamines. The lowest oxidation potential corresponds to EDOT derivative, followed by furan, thiophene and pyridine ones. Increase of this value in presented sequence may be attributed to the rising aromatic character of the side rings, as indicated by their aromatic resonance energy (furan 68 kJ mol−1, pyridine 117 kJ mol−1, thiophene 122 kJ mol−1).[Citation48–51] The presence of pyridine ring hampers oxidation process, significantly rising the oxidation potentials for these molecules. It reflects π-electron-deficient nature of the pyridine ring containing the electron-withdrawing nitrogen atom.[Citation52]
Table 1. Oxidation potentials for investigated monomers and their polymers in 0.1 M TBABF4 in ACN.
The diphenylamine derivatives require less positive potential to gain first oxidation stage in comparison to the triphenylamine ones. In the radical cations, the rings shall change position to allow the π-MOs to become coplanar with the p-orbital at the central nitrogen. The additional steric and electronic interactions caused by the third phenyl ring hamper removal of electron. Increase in potential leads to further oxidation of molecules, which is a multistep process.
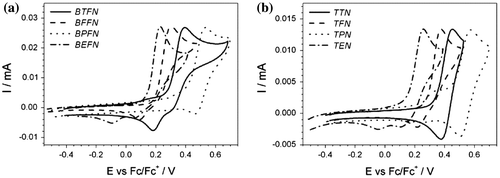
Repetitive sweeping of the monomer solution with the required potential range produces a uniform film on the platinum electrode surface (Figure (a)–(d)). This is the case for each monomer excluding BPFN and TPN. Multiple CV scans reveal changes in CV curves with additional peaks at lower potentials on both the cathodic and anodic scans. This suggests electrochemical coupling at the side ring, indicating electrochemically nonstable molecules. In oxidized monomers, delocalization of charge between the central amine and tienyl, furyl or EDOT side groups occurs. That enables the oxidative polymerization. This process proceeds through coupling at the unsubstituted (α) positions of the side ring. Stable layers were deposited on both Pt and ITO electrode; however, it was easier to start the nucleation process on the Pt surface and hence more compact layers were formed.
Figure 4. Cyclic voltammetry of (a) BTFN, (b) TTN, (c) BEFN and (d) TEN in 0.1 M solution of TBABF4 in ACN. Potential sweep rate v = 0.050 V s−1.
The potential required to start electropolymerization was assessed on the basis of CV experiment . The applied upper potential boundary was close to the first oxidation peak of the monomer. Growth of the current value in successive cycles reflected synthesis of conducting layer on the working electrode surface. For monomers where no such growth was obtained, a higher potential value was applied until the layer could be deposited. The potential required to start continuous growth of the polymer is close to the first oxidation peak for furan (BFFN, TFN) and EDOT (BEFN and TEN) derivatives. For thiophene derivatives this potential must be higher, close to the second oxidation peak. Potential of oxidation for furan and EDOT rings are close enough to the oxidation potential for phenylamine part, so these two processes take place almost simultaneously. Electropolymerizations of BTFN and TTN were successful after cycling beyond the second oxidation stage of monomers. It suggests that the first oxidation processes for these monomers are based largely on the amine centre while the second oxidations are taking place at the thienyl part. For furyl and EDOT derivatives, charge is more delocalized between the central amine part and side rings.
Redox response of polymers
When the as-prepared polymers of polydiphenylamines and polytriphenylamines (Figure (a)–(d) were transferred into an electrolyte solution without the monomer, voltammetric curves were recorded (Figure ). The shapes of the polymer curves match very well to those obtained during the process of polymerization. This fact suggests the quantitative polymerization of monomers occurred with small amounts of possible by-products. The polymeric films obtained in repetitive CV experiments were examined in doping and dedoping process in the monomer-free solutions. Table contains the oxidation potential for the deposited films. All of them were yellow and during doping process, their colour was changed into blue or blanched, but dedoping process restored the colour in a reversible way.
Figure 5. CV of (a) polydiphenylamine and (b) polytriphenylamine polymer films in 0.1 M solution of TBABF4 in ACN. Potential sweep rate v = 0.050 V s−1.
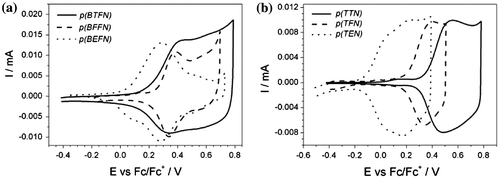
The CVs of the polymers exhibit similar features with dominant oxidation waves peaking at 0.406, 0.275 and 0.145 V for diphenylamines (p(BTFN), p(BFFN), p(BEFN), respectively) (Figure (a)) and 0.558, 0.404 and 0.058 V for triphenylamines (p(TTN), p(TFN), p(TEN), respectively) (Figure (b)). Parent P(DPN) undergoes redox transitions at 0.337 V vs Fc/Fc+,[Citation43] 0.35 and 0.73 V vs Fc/Fc+ (in 0.1 M [Et4 N]BF4 in ACN),[Citation53] 0.337 V vs Fc/Fc+ (in 0.1 M LiClO4/ACN) [Citation54] while p(TPN) – at 0.42 V vs Fc/Fc+ (Pt electrode, 0.1 M Bu4NPF6/ACN),[Citation55] 0.44 V vs Fc/Fc+.[Citation36] Hence, the first redox pair is assigned to represent the redox transitions of phenylamine units within the polymer chain. In diphenylamine group, first redox system dominates over the second one and they are better separated. For diphenylamine series, the two waves reflect oxidation process for the amine and the biheterocyclic part of the polymer chains. Due to the presence of either bithiophene, bifurane or biEDOT units in the polymer, redox reactions connected with these conjugated units may also occur. For triphenylamine series, the biheterocyclic and amine parts are also oxidized. However, the presence of broad oxidation wave suggests that in this series, the two oxidations occur at the same time. It may also suggest large diversity of conjugation length for deposited polymers.
For the oxidation process, the onset potential value corresponds to the highest occupied states in the valence band [Citation56,57] and no charges can be removed from the polymer until the applied potential reaches this value.[Citation58] In the present study, the onset of the oxidation potential was lower for polymers derived from derivatives of diphenylamines. It implies that for this group it was possible to deposit chains with longer effective conjugation length, or in the structure of the material were present ordered areas. It was triphenylamine group though that was more stable during repetitive potential sweeping. In all cases, the smooth solid films were obtained with the polydiphenylamines being more uniformly dispersed on the electrode surface. No sign of film cracking or detachment was observed during doping and dedoping cycles, which indicates high film durability.
Thin-layer polymerization
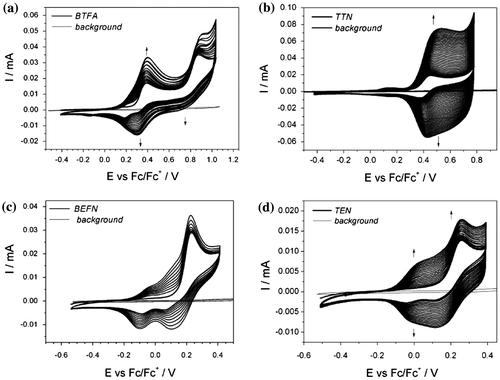
Polymerization process was traced during thin-layer measurement when the increasing potential was applied and corresponding UV–vis spectra were collected (Figure ). Spectrum of cleaned ITO slide was recorded first to obtain a background scan. It was further abstracted from the spectra recorded for investigated sample.
Figure 6. UV–vis spectrum recorded during spectroelectrochemical experiments for (a) BTFN, (b) TTN in 0.1 M TBABF4 in ACN.
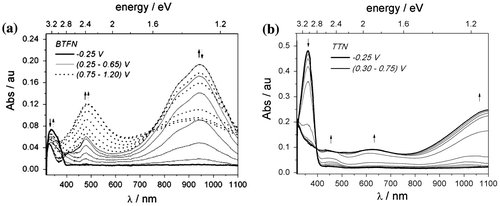
At the beginning of the measurement, there is a single absorption band characteristic to monomer molecule for diphenylamine ( = 338 nm (BTFN), 349 nm (BFFN), 360 nm (BPFN), 362 nm (BEFN)) and triphenylamine derivative (
= 361 nm (TTN), 355 nm (TFN), 360 nm (TPN), 372 nm (TEN)). Increasing the potential leads to oxidation process as electron is removed from the neutral monomer molecule. It is reflected as a decrease in intensity of the monomer’s absorption band. At the same time, new bands are being formed (λ1 = 400–550 nm, λ2 = 700–1100 nm) which confirms formation of other reactive species (radical cation). For BTFN molecule, application of higher potential causes further increase in intensity for band located in the region λ1 = 400–550 nm, while the intensity of the second band (located in the region λ2 = 700–1100 nm) is markedly diminished.
The shape of the UV–vis spectrum is similar for three diphenylamine derivatives, with two distinctive regions – very intensive low-energy band located in the range (700–1100) nm and relatively sharp peak in the range (400–550) nm. Their presence confirms generation of respective phenylamine radical cations. The formation of one well-defined isobestic point for all derivatives indicates coexistence of two phases during this transition. After reverse of polarization direction in measurement cell, the shape of the spectrum is changed. Newly formed bands indicate the presence of longer chain as can be deduced on the bathochromic shift of absorption bands. For thiophene derivative, the shape of the spectrum changes considerably after application of higher voltage corresponding to the second oxidation stage. It results in decrease intensity of low-energy band with simultaneous increase in intensity for band located at (400–550) nm. Well-defined isobestic point is formed at 747 nm and a little blue-shift in the band location is observed.
Results of spectroelectrochemical measurements for triphenylamine derivatives (Figures (b)) reveal the presence of newly formed bands corresponding to radical cation moieties. However, low-energy bands position could not be determined as they move to IR part of the spectrum and only their slopes were detected within the applied measurement scope. Only for furan derivative it was possible to record the spectrum, which implies shorter conjugation range for the reaction product. In comparison to diphenylamine derivatives, the intensity of the newly forming bands is lower. For pirydyl monomers (BPFN and TPN), the system is not reversible – the electron transfer reaction at the electrode induces chemical reaction and after application of the reverse potential, the spectrum returns to the original position but not to the original intensity.
Photophysical studies of polymers
Polymeric films for spectroelectrochemical measurements were deposited on ITO surface in CV experiment. Before recording the UV–vis spectra, all films were dedoped and carefully washed with the solvent (ACN) to remove the monomer and soluble short-chain fraction. Figure presents UV–vis spectra of polymeric films. The spectra of the dedoped films are similar in shape with the position of the absorption maximum located at 428, 380, 445 nm for p(BTFN), p(BFFN), p(BEFN) and 432, 388 and 448 nm for p(TTN), p(TFN) and p(TEN), respectively (Table ). The absorption band, due π−π* transitions in alternately conjugated polymers, is always shifted to lower energies with respect to monomers. Its shape represents distribution lengths of conjugated segments on various polymer chains within thin film volume. The data imply that the effective conjugation length is longer for triphenylamines than for diphenylamines. The influence of the side ring is also visible. The location of π−π* absorption bands points that in case of furan derivatives, the conjugation length is the shortest, increasing with thiophene substitution and becoming the longest in case of EDOT derivatives. Furan ring is known from its susceptibility for photooxidation,[Citation59,60] which might be the reason for this short conjugation. For EDOT derivatives, the conjugation is the longest as these molecules undergoes oxidation at the lowest potential, reducing the risk of overpotential degradation. Additionally, the shoulder observed in the spectrum suggests a rigidification of the conjugated branches by intramolecular sulphur-oxygen interactions as reported for EDOT-containing systems.[Citation60,61] The absorption bands for all investigated polymers are broad indicating large diversity in effective conjugation length of polymer chains.
Figure 7. UV–vis spectra of dedoped polymeric films in 0.1 M TBABF4 in ACN.
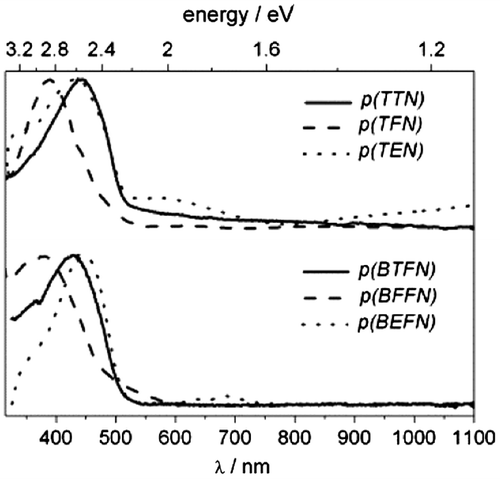
Table 2. Absorption maximums (λmax) and energy gap values ( ) for investigated polymers; was estimated from onset of π−π* absorption band ( = h·c/λonset).
) for investigated polymers; was estimated from onset of π−π* absorption band ( = h·c/λonset).
Figure presents the evolution of the absorption spectrum of thin films upon electrochemical doping. Increasing the applied potential and hence the doping level produces a decrease of the intensity of the π−π* transition and hypsochromical shift of absorption maximum. Concurrently, two new broad absorption features appear in the region at (500–800) nm and above 900 nm. These absorption bands can be attributed to electronic transitions between the valence band and polaron level. The absorbance variations occur at well-defined isosbestic point at 500 nm, indicating that only two phases coexist in the system.
Figure 8. UV–vis spectrum recorded during spectroelectrochemical experiments for (a) p(BTFN), (b) p(TTN) in 0.1 M TBABF4 in ACN.
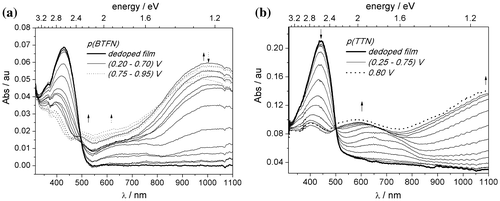
The width of the energy gaps (Eg) for dedoped films were determined from the low-energy onset of the absorption bands (Table ). The values are lower in case of triphenylamine derivatives and the presence of EDOT unit.
The additional electron donor group’s effect on HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) levels was also investigated. The energy diagram is shown in Figure . HOMO and LUMO levels are estimated by using the equations EHOMO = −(Eox + 4.8) eV and Eg = EHOMO − ELUMO, where Eox is the onset oxidation potential regarding the standard ferrocene/ferrocenium (Fc/Fc+) redox system.
Figure 9. HOMO–LUMO energy levels for obtained polymers.
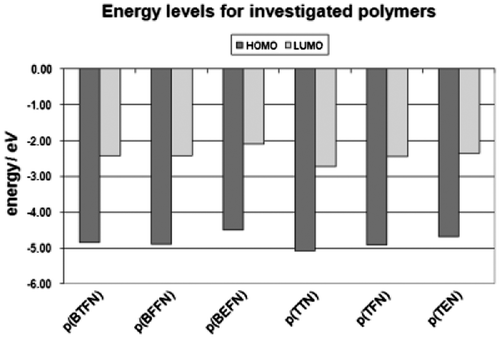
In diphenylamine group, the Eg value of furane derivative is slightly higher than for thiophene one (2.49 and 2.42 eV, respectively), while the presence of EDOT rings lowers this value (2.40 eV). This order is the same for triphenylamine group. However, additional phenyl ring allows to reduce Eg value by both increase in energy of HOMO level and decrease in energy of LUMO level of polymers.
Morphological analysis
The morphology of polymers was investigated by taking tapping-mode AFM images of ultrathin films on ITO slides. Figure shows typical AFM images of the polymer-coated electrodes. The polymer is obtained as granules (size 100–200 nm), which form larger agglomerates.
Figure 10. Tapping mode AFM images of polydiphenylamines and polytriphenylamines films on ITO slides: (a) p(BTFN), (b) p(TTN), (c) p(BFFN), (d) p(TFN), (e) p(BEFN) and (f) p(TEN); scan range: 20x20 μm.
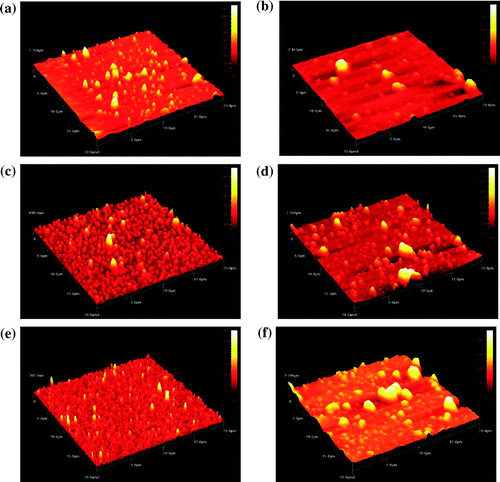
For polydiphenylamines, the AFM micrograph (Figure (a), (c), and (e)) reveals more homogeneous structure with many spike structures. For polytriphenylamines (Figure (b), (d), and (f)), the technique shows more diversity in size of clusters. It is observed that a surface reveals less compact morphology with large aggregates with diameters up to 5.0 μm.
Conclusions
Based on the obtained results, we state that electroactivity of monomers with phenylamine core and heterocyclic side groups allows them to grow into polymer chains. It was shown that heterocyclic side rings influence markedly on the molecule oxidation potential and hence on the ionization potential. The decrease of the value below the oxidation potential for parent phenylamine units is the outcome of this behaviour. The removal of electron is easier for diphenylamine derivatives than for the triphenylamine ones. The reason for that might be steric hindrance encountered when the third phenyl ring occupies the position in the neighbourhood of the nitrogen atom.
Enlarged conjugated system in triphenylamine-based polymer influences their stability. The position of the side ring controls the polymer structure and hence the mean conjugation length within the macromolecules. Introduction of additional branch into the phenylamine monomer structure allows to lower the energy gap value of the final polymer. However, it increases oxidation potential at the same time.
AFM micrograph of polydiphenylamines reveals more homogeneous structure with many spike structures, while morphology of polytriphenylamines is less compact with large aggregates with diameters up to 5.0 μm.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Inzelt G, Pineri M, Schultze JW, Vorotyntsev MA. Electron and proton conducting polymers: recent developments and prospects. Electrochim. Acta. 2001;45:2403–2421.
- Sariciftci NS, Kuzmany H, Neugebauer H, Neckel A. Structural and electronic transitions in polyaniline: a Fourier transform infrared spectroscopic study. J. Chem. Phys. 1990;92:4530–4539.10.1063/1.457765
- Camalet JL, Lacroix JC, Aeiyach S, Chane-Ching K, Lacaze PC. Electrosynthesis of adherent polyaniline films on iron and mild steel in aqueous oxalic acid medium. Synth. Met. 1998;93:133–142.10.1016/S0379-6779(97)04099-X
- Salaneck SW, Huang WS, Lundstrom I, Macdiarmid AG. A two-dimensional-surface ‘state diagram’ for polyaniline. Synth. Met. 1986;13:291–297.10.1016/0379-6779(86)90190-6
- Guay J, Dao LH. Formation of poly(4-phenylaniline) by electropolymerization of 4-aminobiphenyl or diphenylamine. J. Electroanal. Chem. 1989;274:135–142.10.1016/0022-0728(89)87035-4
- Hayat U, Dodd PN, Baker GH. Electrochemical synthesis and study of polydiphenylamine. J. Electroanal. Chem. Interfacial Electrochem. 1987;220:287–294.10.1016/0022-0728(87)85115-X
- Yang CY, Smith P, Heeger A, Cao Y, Osterholm JE. Electron diffraction studies of the structure of polyaniline-dodecylbenzenesulfonate. Polymer. 1994;35:1142–1147.10.1016/0032-3861(94)90004-3
- Athawale AA, Deore BA, Chabukswar VV. Studies on poly(diphenylamine) synthesized electrochemically in nonaqueous media. Mater. Chem. Phys. 1999;58:94–100.10.1016/S0254-0584(98)00258-2
- Comisso N, Daolio S, Mengoli G, Salmaso R, Zecchin S, Zotti G. Chemical and electrochemical synthesis and characterization of polydiphenylamine and poly-N-methylaniline. J. Electroanal. Chem. Interfacial Electrochem. 1988;255:97–110.10.1016/0022-0728(88)80007-X
- de Santana H, Temperini MLA, Rubim JC. In situ resonance Raman and reflectance spectroscopic study of the electrochemical oxidation of diphenylamine. J. Electroanal. Chem. 1993;356:145–155.10.1016/0022-0728(93)80516-K
- Chung CY, Wen TC, Gopalan A. Identification of electrochromic sites in poly(diphenylamine) using a novel absorbance–potential–wavelength profile. Electrochim. Acta. 2001;47:423–431.10.1016/S0013-4686(01)00742-3
- Kunugi Y, Tabakovic I, Canavesi A, Miller L. Light-emitting diodes based on linear and starburst electro-oligomerized thienyltriphenylamines. Synth. Met. 1997;89:227–229.10.1016/S0379-6779(97)81223-4
- Tang CW, VanSlyke SA. Organic electroluminescent diodes. Appl. Phys. Lett. 1987;51:913–915.10.1063/1.98799
- Chang S-C, Liu J, Bharathan J, Yang Y, Onohara J, Kido J. Multicolor organic light-emitting diodes processed by hybrid inkjet printing. Adv. Mater. 1999; 11: 734–737.10.1002/(ISSN)1521-4095
- Bach U, De Cloedt K, Spreitzer H, Grätzel M. Characterization of hole transport in a new class of spiro-linked oligotriphenylamine compounds. Adv. Mater. 2000;12:1060–1063.10.1002/(ISSN)1521-4095
- Mitschke U, Osteritz EM, Dedaerdemaeker T, et al. A novel series of p−n diblock light-emitting copolymers based on oligothiophenes and 1,4-Bis(oxadiazolyl)-2,5-dialkyloxybenzene. Macromolecules. 1999;32:118–126.
- Kuwabara Y, Ogawa H, Inada H, Noma N, Shirota Y. Thermally stable multilared organic electroluminescent devices using novel starburst molecules, 4,4′,4″-Tri(N-carbazolyl)triphenylamine (TCTA) and 4,4′,4″-Tris(3-methylphenylphenylamino)triphenylamine (m-MTDATA), as hole-transport materials. Adv. Mater. 1994;6:677–679.10.1002/(ISSN)1521-4095
- Tanaka S, Takeuchi K, Asai M, Iso T, Ueda M. Preparation of hyperbranched copolymers constituted of triphenylamine and phenylene units. Synth. Met. 2001;119:139–140.10.1016/S0379-6779(00)01268-6
- Yamamoto K, Higuchi M, Uchida K, Kojima Y. Synthesis and electrochemical properties of oligo- and poly(thienylphenylamine)s. Macromolecules. 2002;35:5782–5788.10.1021/ma011018c
- Kvarnström C, Petr A, Damlin P, Lindfors T, Ivaska A, Dunsch L. Raman and FTIR spectroscopic characterization of electrochemically synthesized poly(triphenylamine), PTPA. J. Solid State Electrochem. 2002;6:505–512.10.1007/s10008-002-0275-6
- Sim J, Ueno E, Natori I, Ha J, Sato H. Oxidation polymerization of N-butyl-N,N-diphenylamine (BDPA) and N-4-butylphenyl-N,N-diphenylamine (BTPA). Synth. Met. 2008; 158: 345–349.10.1016/j.synthmet.2008.02.005
- Bui T-T, Beouch L, Sallenave X, Goubard F. Carbazol-N-yl and diphenylamino end-capped triphenylamine-based molecular glasses: synthesis, thermal, and optical properties. Tetrahedron Lett. 2013;54:4277–4280.10.1016/j.tetlet.2013.05.152
- Idzik K, Sołoducho J, Łapkowski M, Golba S. Development of structural characterization and physicochemical behaviour of triphenylamine blocks. Electrochim. Acta. 2008;53:5665–5669.10.1016/j.electacta.2008.03.019
- Idzik K, Sołoducho J, Cabaj J, Mosiądz M, Łapkowski M, Golba S. Novel aspects of a convenient synthesis and of electroproperties of derivatives based on diphenylamine. Helv. Chim. Acta. 2008;91:618–627.10.1002/(ISSN)1522-2675
- Wang B-C, Liao H-R, Chang J-C, Chen L, Yeh JT. Electronic structure and molecular orbital study of hole-transport material triphenylamine derivatives. J. Lumin. 2007; 124: 333–342.10.1016/j.jlumin.2005.11.016
- Nelson RF, Adams RN. Anodic oxidation pathways of substituted triphenylamines. II. Quantitative studies of benzidine formation. J. Am. Chem. Soc. 1968;90:3925–3930.10.1021/ja01017a004
- Fehér K, Inzelt G. Electrochemical quartz crystal microbalance study of formation and redox transformations of poly(diphenylamine). Electrochim. Acta. 2002;47:3551–3559.10.1016/S0013-4686(02)00359-6
- Inzelt G. Cyclic voltammetry of solid diphenylamine crystals immobilized on an electrode surface and in the presence of an aqueous solution. J. Solid State Electrochem. 2002;6:265–271.10.1007/s100080100223
- Seo ET, Nelson RF, Fritsch JM, Marcoux LS, Leedy DW, Adams RN. Anodic oxidation pathways of aromatic amines. Electrochemical and electron paramagnetic resonance studies. J. Am. Chem. Soc. 1966;88:3498–3503.10.1021/ja00967a006
- Wang H-Y, Chen L-F, Zhu X-L, Wang Ch, Wan Y, Wu H. Spectral studies of multi-branched fluorescence dyes based on triphenylpyridine core. Spectrochim. Acta, Part A. 2014;121:355–362.10.1016/j.saa.2013.10.087
- Wu G, Kong F, Li J, et al. Triphenylamine-based organic dyes with julolidine as the secondary electron donor for dye-sensitized solar cells. J. Power Sources. 2013;243:131–137.10.1016/j.jpowsour.2013.05.188
- Yang P-C, Wu H, Lee C-L, Chen W-C, He H-J, Chen M-T, Triphenylamine-based linear conjugated polyfluorenes with various pendant groups: synthesis, characterization, and ion responsive properties. Polymer. 2013;54:1080–1090.10.1016/j.polymer.2012.12.071
- Kochapradist P, Prachumrak N, Tarsang R, et al. Multi-triphenylamine-substituted carbazoles: synthesis, characterization, properties, and applications as hole-transporting materials. Tetrahedron Lett. 2013;54:3683–3687.10.1016/j.tetlet.2013.05.007
- Shen S, Gao L, He Ch, Zhang Z, Sun Q, Li Y. A star-shaped oligothiophene with triphenylamine as core and octyl cyanoacetate as end groups for solution-processed organic solar cells. Org. Electron. 2013;14:875–881.10.1016/j.orgel.2012.12.030
- Chen S, Gao Q, Zhao J, Cui C, Yang W, Zhang X. Electrochemical Synthesis and Characterization of a new electrochromic copolymer based on 2,2'-bithiophene and tris[4-(2-Thienyl)phenyl]amine. Int. J. Electrochem. Sci. 2012;7:9095–9112.
- Chahma M, Gilroy JB, Hicks RG. Linear and branched electroactive polymers based on ethylenedioxythiophene–triarylamine conjugates. J. Mater. Chem. 2007;17:4768–4771.10.1039/b711693d
- Cremer J, Briehn CA. Novel highly fluorescent triphenylamine-based oligothiophenes. Chem. Mater. 2007;19:4155–4165.10.1021/cm0700448
- Luponosov YN, Solodukhin AN, Ponomarenko SA. Branched triphenylamine-based oligomers for organic electronics. Polym. Sci. Ser. C. 2014;56:104–134.10.1134/S181123821401007X
- Sek D, Iwan A, Jarzabek B, et al. Hole transport triphenylamine−azomethine conjugated system: synthesis and optical, photoluminescence, and electrochemical properties. Macromolecules. 2008;41:6653–6663.10.1021/ma702637k
- Sęk D, Lapkowski M, Dudek H, Karoń K, Janeczek H, Jarząbek B. Optical and electrochemical properties of three-dimensional conjugated triphenylamine-azomethine molecules. Synth. Met. 2012;162:1046–1051.10.1016/j.synthmet.2012.04.024
- Park H, Oyama M. Remarkable 3-methyl substituent effects on the cyclization reaction of diphenylamine derivative cation radicals in acetonitrile. J. Chem. Soc., Perkin Trans. 2. 2002;7:1335–1339.10.1039/b201796b
- Oyama M, Imabayashi T, Ho J-H, Ho T-I. Reaction analysis of 3-substituted-diphenylamine cation radicals in acetonitrile. Cyclization reaction vs. benzidine formation. Electrochemistry. 2006;74:649–655.
- Wang YJ, Sheu HS, Lai CK. New star-shaped triarylamines: synthesis, mesomorphic behavior, and photophysical properties. Tetrahedron. 2007;63:1695–1705.10.1016/j.tet.2006.11.058
- Creason SC, Wheeler J, Nelson RF. Electrochemical and spectroscopic studies of cation radicals. I. coupling rates of 4-substituted triphenylaminium ion. J. Org. Chem. 1972;37:4440–4446.10.1021/jo00799a034
- Quinton C, Alain-Rizzo V, Dumas-Verdes C, Miomandre F, Clavier G, Audebert P. Redox-controlled fluorescence modulation (electrofluorochromism) in triphenylamine derivatives. RSC Adv. 2014;4:34332–34342.10.1039/C4RA02675F
- Cremer J, Bäuerle P. Star-shaped perylene–oligothiophene–triphenylamine hybrid systems for photovoltaic applications. J. Mater. Chem. 2006;16:874–884.10.1039/B515657B
- Pacansky J, Waltman RJ, Seki H. Ab initio computational studies on the structures and energetics of hole transport molecules: triphenylamine. Bull. Chem. Soc. Japan. 1997;70:55–59.10.1246/bcsj.70.55
- Newkome GR, Paudler WW. Contemporary heterocyclic chemistry. New York (NY): Wiley; 1982.
- Kumagai A, Fukumoto H, Yamamoto T. Chemical and electrochemical oxidation of thiophene−pyridine and thiophene−pyrimidine co-oligomers in solutions. J. Phys. Chem. B. 2007;111:8020–8026.10.1021/jp071614u
- Jenkins IH, Salzner U, Pickup PG. Conducting copolymers of pyridine with thiophene, N-methylpyrrole, and selenophene. Chem. Mater. 1999;8:2444–2450.
- Yamamoto T, Zhou ZH, Kanbara T, et al. π-conjugated donor−acceptor copolymers constituted of π-excessive and π-deficient arylene units. Optical and electrochemical properties in relation to CT structure of the polymer. J. Am. Chem. Soc. 1996;118:10389–10399.10.1021/ja961550t
- Yamamoto T, Maruyama T, Zhou ZH, et al. pi.-conjugated poly(pyridine-2,5-diyl), poly(2,2'-bipyridine-5,5'-diyl), and their alkyl derivatives. Preparation, linear structure, function as a ligand to form their transition metal complexes, catalytic reactions, n-type electrically conducting properties, optical properties, and alignment on substrates. J. Am. Chem. Soc. 1994;116:4832–4845.10.1021/ja00090a031
- Kim S-B, Harada K, Yamamoto T. Preparation of poly(diphenylamine-4,4′-diyl) and related random copolymers by organometallic polycondensation. Electrical, electrochemical, and optical properties. Macromolecules. 1998;31:988–993.10.1021/ma971244f
- Guay J, Paynter R, Dao LH. Synthesis and characterization of poly(diarylamines): a new class of electrochromic conducting polymers. Macromolecules. 1990;23:3598–3605.10.1021/ma00217a010
- Petr A, Kvarnström C, Dunsch L, Ivaska A. Electrochemical synthesis of electroactive polytriphenylamine. Synth. Met. 2000;108:245–247.10.1016/S0379-6779(99)00137-X
- De Leeuw DW, Simenon MMJ, Brown AR, Einerhand REF. Stability of n-type doped conducting polymers and consequences for polymeric microelectronic devices. Synth. Met. 1997;87:53–59.10.1016/S0379-6779(97)80097-5
- Pommerehne J, Vestweber H, Guss W, Mahrt RF, et al. Efficient two layer leds on a polymer blend basis. Adv. Mater. 1995;7:551–554.10.1002/(ISSN)1521-4095
- Micaroni L, Nart FC, Hümmelgen IA. Considerations about the electrochemical estimation of the ionization potential of conducting polymers. J. Solid State Electrochem. 2002;7:55–59.
- Saadeh H, Goodson T, Yu L. Synthesis of a polyphenylene-co-furan and polyphenylene-co-thiophene and comparison of their electroluminescent properties. Macromolecules. 1997;30:4608–4612.10.1021/ma970577+
- Leriche P, Turbiez M, Monroche V, Frère P, Blanchard PJ, Skabara PJ. Strong π-electron donors based on a self-rigidified 2,2′-bi(3,4-ethylenedioxy)thiophene–tetrathiafulvalene hybrid π-conjugated system. Tetrahedron Lett. 2003;44:649–652.10.1016/S0040-4039(02)02702-8
- Cravino A, Roquet S, Alévêque O, Leriche P, Frère P, Roncali J. Triphenylamine−oligothiophene conjugated systems as organic semiconductors for opto-electronics. Chem. Mater. 2006;18:2584–2590.10.1021/cm060257h