
ABSTRACT
Background
The α-thalassemia is a highly prevalent disease in tropical and subtropical regions, including southern China, and is mainly caused by deletion in α-globin genes (HBA1 and HBA2). The clinical manifestation of α-thalassemia is highly correlated with the copy number of α-globin genes. The decrease in copy number results in α-thalassemia, while duplication or triplication compounded with β-thalassemia may aggravate the clinical manifestation. However, the common methods used to measure the copy number variants can only detect the three common types: –SEA, -α3.7, and -α4.2, and may easily miss the rare deletional type and duplication or triplication cases. Therefore, a new method that allows the detection of different copy number variants in α-globin genes simultaneously and accurately needs to be established.
Methods
A total of 428 peripheral-blood and fetal chorionic villus or amniotic fluid samples were used in this study. We employed a pair of primers and two probes, one for HBA1 and another for HBA2, to perform droplet digital polymerase chain reaction (ddPCR). Each reaction needed the ddPCR of RPP30 as a reference gene to calculate the copy number.
Results
We accurately detected the copy number variants in α-globin genes, including the common form α-thalassemia, triplications such as αααanti4.2, and trisomy 16, by performing only two reactions. The accuracy rate for detecting the copy number of α-globin genes was up to 100%.
Conclusion
In conclusion, ddPCR served as an accurate and rapid method for detecting copy number variations in the clinical screening for α-thalassemia.
Introduction
The α-thalassemia is one of the most common inherited disorders worldwide [Citation1]. It mainly occurs in tropical and subtropical regions, including southern China, with a high carrier rate of 11.31% and 17.55%, respectively, in Guangdong and Guangxi provinces [Citation2,Citation3]. It is due to the deletion or non-deletion in the α-globin cluster, which is located on chromosome 16. The α-globin cluster has seven genes arranged as follows: 5´-ς2-Ψς1-Ψα2-Ψα1-α2-α1-θ-3´. α2 (HBA2) and α1 (HBA1) are highly homologous, except for the sequences in +55 and +119 sites in the second intron; the sequences are T and G in HBA2 and G and CTCGGCCC in HBA1 in +55 and +119 sites, respectively. The clinical manifestations of α-thalassemia mainly depend on mutations in the α-globin cluster. The most common α-thalassemia mutations in southern China are caused by deletion in α-globin genes, including –SEA, -α3.7, and -α4.2. The homozygosity of –SEA results in Hb Bart’s hydrops fetalis and leads to death during gestation or in several hours after birth, whereas –SEA deletion heterozygosity compounded with -α3.7 or -α4.2 deletion causes Hb H disease, in which the patient may depend on irregular blood transfusion to survive. The triplication of α-globin, such as αααanti3.7 and αααanti4.2, compounded with β-thalassemia, may aggravate the clinical manifestation, which was also reported in a Chinese population [Citation4]. In addition, the carrier rate of triplications and quadruplications of α-globin is relatively not low, about 2% in southern China [Citation5]. Therefore, detecting the copy number variation (CNV) of α-globin genes while one of the parents or both are β-thalassemia carriers in prenatal diagnosis is of significance.
The main methods used for detecting CNV in α-globin genes include multiplex gap-polymerase chain reaction (PCR) [Citation6], multiplex ligation-dependent probe amplification (MLPA) [Citation7], real-time quantitative PCR (qPCR) [Citation8,Citation9], and so on. However, these methods have certain limitations. Although the multiplex gap-PCR is relatively less expensive and does not require high-level equipment, it can only detect the known common deletional mutations. The variation types of α-globin gene copy number are complex and diverse, and multiple gap-PCR inevitably leads to missed diagnosis. MLPA is mainly used for the detection and genotype confirmation of CNVs of unknown types in individual cases because of its complicated operation, high time consumption, and high cost. QPCR is needed to establish standard curves and CT values for quantitative copy number analysis, rather than for direct quantitative analysis.
Droplet digital PCR (ddPCR), with high throughput and easy operation, can accurately and directly detect CNV without standard curves and CT values. It stands out in CNV detection due to the use of microfluidic technology to separate each reaction mix into thousands of micro-spherical droplets [Citation10]. The detection of –SEA deletion using ddPCR was established in 2013 by Watcharee Prasing from Thailand [Citation11]. This was the first time for using ddPCR in α-thalassemia diagnosis; however, it could only detect –SEA deletion. In 2016, Elizabeth George from Malaysia used ddPCR for detecting triplications and deletions of α-globin genes [Citation12], in which four CNV assays were performed in duplications for each sample, which included HS-40, HBA2, HBA3.7, and HBA1 genes. Although these assays could accurately detect triplications and deletions of α-thalassemia, they were expensive and had reduced throughput. In this study, we established a new method using ddPCR with only two PCR reactions to detect the CNVs of α-globin genes, which included the detection of deletions and triplications.
Methods
Dna samples
A total of 428 DNA samples with characterized genotype were used in this study, including 385 heterozygous and homozygous or compounded heterozygous samples of common deletional α-thalassemia, 25 normal samples, a sample of rare deletional α-thalassemia –THAI compounded with -α3.7, 2 samples with αααanti4.2 heterozygosity, and 14 samples with trisomy 16. One sample with –SEA deletion compounded with HKαα was also enrolled in this study. Informed consent was obtained from all the participants prior to the study following the presentation of the nature of the procedures. The study was conducted following the Declaration of Helsinki.
Droplet digital PCR
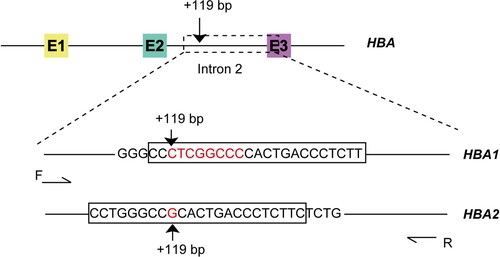
Two PCR reactions with a pair of primers and two probes specific to HBA1 and HBA2 were performed for detecting the CNV of α-globin genes. The locations of the primers and probes are shown in . The ddPCR was conducted following the protocol recommended by the manufacturer (ddPCR Supermix for probes, 1863024; Bio-Rad, Hercules, CA, USA). Each reaction contained 250 nM 1 × ddPCR Supermix for probes (no dUTP). The primers and probes were as follows: primer F: 5´-GGTTGCGGGAGGTGTAGC-3´, primer R: 5´-GTGCTCACAGAAGCCAGGAAC-3´; HBA1 probe (specific to HBA1 gene detection assay): 5´- FAM-CCCTCGGCCCCACTGACCCTCTT-BHQ1-3´; HBA2 probe (specific to HBA2 gene detection assay): 5´- FAM-CCTGGGCCGCACTGACCCTCTTC-BHQ1-3´. RPP30 served as the reference gene, and the specific primers and probe (RPP30 F: 5´- GATTTGGACCTGCGAGC-3´; RPP30 R: 5´-GGTTGGCCAGGCGCGAAG-3´; probe: 5´-VIC-CTGACCTGAAGGCTCT-MGB-3´) were also added to each reaction. Furthermore, 5–10 ng DNA samples were added as the template. Ultrapure water was added up to 20 μL for each reaction. A total of 20 μL of the reaction mix was loaded into a sample well of a DG8-cartridge for QX200 Droplet Generator (Bio-Rad), followed by 70 μL of droplet generator oil into the oil wells. After the droplets were generated, they were carefully transferred into a clean 96-well plate. The plate was sealed using the PX1 PCR Plate Sealer at 180°C for 5 s. Then, thermal cycling was performed as follows: 25°C hold for 3 min, 95°C for 10 min; then 40 cycles at 94°C for 30 s and 61°C for 1 min. After 40 cycles, the plate was incubated at 98°C for 10 min and held at 4°C. After thermal cycling, the plate was transferred to the QX200 Droplet Reader for data acquisition.
Figure 1. Schematic of HBA genes. E1–E3, exon 1–3. The black boxes show the probes specific to HBA1 and HBA2.
Cnv analysis
To calculate the copy number, we used RPP30 (which had two copy numbers) as the reference gene. The copy number was calculated as follows: CNV = (X/Y)Nref, where X = the concentration of target genes, Y = the concentration of RPP30, and Nref = the copy number of RPP30 (usually 2). The error bars were 95% confidence interval of Poisson distribution.
Statistical analysis
Data were expressed as the mean ± standard deviation.
Results
We used a pair of primers and two probes specific to HBA1 and HBA2 to perform ddPCR so as to detect the CNV of α-globin genes in deletional α-thalassemia. We found that ddPCR could accurately detect the CNVs in α-globin genes (A and ). A total of 25 normal individuals with all intact α-globin genes had a copy number ratio of close or equal to 2 in both HBA1 (1.95 ± 0.19) and HBA2 (1.78 ± 0.24). Further, 160 individuals heterozygous for –SEA deletion had only one copy for both HBA1 and HBA2, while 125 heterozygous for -α4.2 or -α3.7 deletions had two copies for HBA1 and one copy for HBA2. One copy for HBA1 and 0 for HBA2 were found in 50 samples with –SEA deletion compounded with -α3.7 or -α4.2. The individual homozygous for –SEA deletion had zero copy for both HBA1 and HBA2 while the individual homozygous for -α3.7 or -α4.2 had two copies for HBA1 and zero copy for HBA2. Two samples with compounded heterozygosity for -α3.7 and -α4.2 had two copies for HBA1 and zero copy for HBA2. We also found that individuals with –THAI compounded with -α3.7 deletion had one copy for HBA1 and zero copy for HBA2 (B). The genotypes of these samples were previously characterized using gap-PCR or MLPA. These data showed that ddPCR could accurately detect the CNVs in deletional α-thalassemia.
Figure 2. Ratio of copy number of HBA1 and HBA2 in different genotypes. (a) Copy number for HBA1 and HBA2 in different deletional α-thalassemia. (b and c) Individuals with –THAI compounded with -α3.7 deletion (b) and heterozygous for αααanti4.2 (c). Blue droplets indicate positive HBA1 or HBA2, while green droplets represent RPP30.
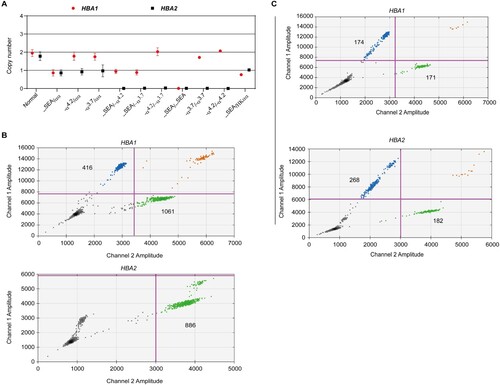
Table 1. Copy number of HBA1 and HBA2 in different genotypes.
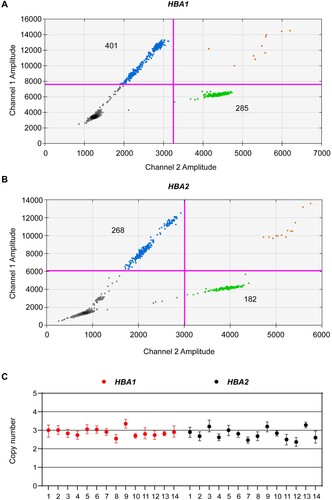
Furthermore, we performed ddPCR by recruiting two individuals with heterozygosity for αααanti4.2 and 14 individuals with trisomy 16 to estimate the accuracy and sensitivity of ddPCR in triplications or multicopy gene detection. We observed that the individuals with heterozygosity for αααanti4.2 had two copies for HBA1 and three copies for HBA2 (C). In addition, the individuals with trisomy 16 had three copies for both HBA1 (2.91 ± 0.21) and HBA2 (2.75 ± 0.45) (). These data indicated that ddPCR could detect the triplications and multicopy genes accurately and sensitively.
Figure 3. DdPCR assay for the quantification of trisomy 16. (a and b) Plots showing the distribution of positive and negative droplets. Blue droplets indicate positive HBA1 or HBA2, while green droplets represent RPP30. (c) Copy number of 14 trisomy 16. Data are shown as mean ± SD.
Discussion
The α-thalassemia, which is mainly caused by a deletion in the α-globin cluster, is one of the most common inherited diseases in tropical or subtropical regions, including southern China. In southern China, the most common α-thalassemia deletions include –SEA, -α3.7, and -α4.2. Therefore, the routine PCR−based method to detect the CNVs mainly focuses only on these three genotypes, but not on triplications such as αααanti3.7 and αααanti4.2, which may lead to misdiagnosis [Citation13]. For example, individuals with HKαα/–SEA are diagnosed as -α3.7/–SEA, but they have α-thalassemia trait hematological data (Hb > 100 g/L) instead of Hb H disease hematological data (Hb < 100 g/L). Considering the carrier rate of HKαα (0.07%) and anti-HKαα (0.02%) in Guangxi province [Citation13], the potential of misdiagnosis cannot be ignored. Also, although the individual with triplications has no clinical manifestation, the symptoms are aggravated when compounded with β-thalassemia. Therefore, to detect triplications and quadruplications, which cannot be detected by using the routine PCR-based method it is very important and meaningful.
To date, many methods, such as MLPA [Citation14,Citation15] and next-generation sequencing (NGS) [Citation5,Citation16] have been developed for CNV analysis. These methods can detect large deletions or unknown deletions, but also have some limitations in clinical screening. MLPA is complicated, time consuming, and expensive. Also, NGS needs high-end equipment and is expensive, which limits its application in the clinical screening of α-thalassemia. DdPCR, which is highly sensitive and time saving, has more advantages compared with other methods. DdPCR requires less time. We could detect the CNVs within 4 h from DNA extraction to data analysis using the method proposed in this study. Furthermore, in this new method, only two reactions were sufficient to cover the detection of deletions and triplications or quadruplications of α-globin genes. Hence, it was more suitable for clinical screening compared with the method reported by Elizabeth George from Malaysia in 2016, in which four CNV assays were performed in duplications for each sample. Furthermore, our purpose was to establish a new method using ddPCR to cover the detection of most of the CNVs of α-globin genes so as to avoid or reduce the misdiagnosis or missed diagnosis in clinical screening and prenatal diagnosis. This new method could accurately and sensitively detect the CNVs of α-globin genes; even only 1 ng DNA could be detected.
However, the use of ddPCR to detect CNVs has some limitations. First, considering only two channels (FAM and HEX or VIC) for the QX200 Droplet Reader, two reactions were required containing probes specific to HBA1 or HBA2 and RPP30 to detect the CNVs. Second, we could detect the CNVs of α-globin genes but could not distinguish the genotypes using this method. For example, it was not possible using this method to be sure about the rare –THAI and –FIL deletions, which appeared the same as –SEA deletion. In these cases, a conventional multiplex gap-PCR might be more attractive. However, although this method could not characterize the exact genotypes, it could be used for primary diagnosis. One individual with Hb H disease will be detected with one copy for HBA1 and zero copy for HBA2. An individual homozygous or compounded heterozygous for -α3.7 or -α4.2 will be detected with two copies for HBA1 and zero copy for HBA2, while the individual heterozygous for -α3.7 or -α4.2 will be detected with two copies for HBA1 and one copy for HBA2. In addition, we could also accurately detect even five or six copies using this new method. Therefore, it was sufficient to use ddPCR to detect CNVs for clinical screening, but other methods such as gap-PCR and MLPA should be used for confirmation.
In summary, we established a new method to detect the CNVs in α-globin genes using a pair of primers and two probes to perform ddPCR in two reactions. This new method was highly sensitive, with an accuracy of up to 100%.
Author contributions
X.B. and J.M. performed the assays, analyzed data and wrote the manuscript; D.Q., X.Z., J.W. and C.Y. collected the samples and performed the confirmed assays. L.Z. and L.D. designed the study and revised the article. All authors reviewed, edited, and approved the version to be submitted.
Availability of data and material
All the data in this study were available in the figures and tables in the manuscript.
Consent to participate and for publication
The patients agreed to participate and be publicized in this study and signed the informed consent.
Ethics approval
This study was approved by the Ethics Committee of Guangdong Women and Children Hospital and was performed in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
Acknowledgments
We thank the patients for their willingness to participate in this study. The financial support from the Science and Technology Program of Guangzhou, China (grant number 202002030390) is gratefully acknowledged.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Higgs DR, Gibbons RJ. The molecular basis of alpha-thalassemia: a model for understanding human molecular genetics. Hematol Oncol Clin North Am. 2010;24:1033–1054. doi:10.1016/j.hoc.2010.08.005.
- Yin A, Li B, Luo M, et al. The prevalence and molecular spectrum of alpha- and beta-globin gene mutations in 14,332 families of Guangdong Province, China. PLoS One. 2014;9:e89855. doi:10.1371/journal.pone.0089855.
- Xiong F, Sun M, Zhang X, et al. Molecular epidemiological survey of haemoglobinopathies in the Guangxi Zhuang Autonomous Region of southern China. Clin Genet 2010;78:139–148. doi:10.1111/j.1399-0004.2010.01430.x.
- Chen W, Zhang X, Shang X, et al. The molecular basis of beta-thalassemia intermedia in southern China: genotypic heterogeneity and phenotypic diversity. BMC Med Genet. 2010;11:31. doi:10.1186/1471-2350-11-31.
- Shang X, Peng Z, Ye Y, et al. Rapid targeted next-generation sequencing platform for molecular screening and clinical genotyping in subjects with hemoglobinopathies. EBioMedicine. 2017;23:150–159. Doi:10.1016/j.ebiom.2017.08.015.
- Liu YT, Old JM, Miles K, et al. Rapid detection of alpha-thalassaemia deletions and alpha-globin gene triplication by multiplex polymerase chain reactions. Br J Haematol. 2000;108:295–299. Doi:10.1046/j.1365-2141.2000.01870.x.
- Gilad O, Shemer OS, Dgany O, et al. Molecular diagnosis of alpha-thalassemia in a multiethnic population. Eur J Haematol. 2017;98:553–562. doi:10.1111/ejh.12866.
- Ke LY, Chang JG, Chang CS, et al. Rapid screening for deleted form of beta-thalassemia by real-time quantitative PCR. J Clin Lab Anal. 2017;31:e22019. doi:10.1002/jcla.22019.
- Tungwiwat W, Fucharoen S, Fucharoen G, et al. Development and application of a real-time quantitative PCR for prenatal detection of fetal alpha(0)-thalassemia from maternal plasma. Ann N Y Acad Sci. 2006;1075:103–107. doi:10.1196/annals.1368.013.
- Kinugasa H, Nouso K, Tanaka T, et al. Droplet digital PCR measurement of HER2 in patients with gastric cancer. Br J Cancer. 2015;112:1652–1655. Doi:10.1038/bjc.2015.129.
- Pornprasert S, Prasing W. Detection of alpha(0)-thalassemia South-East Asian-type deletion by droplet digital PCR. Eur J Haematol. 2014;92:244–248. doi:10.1111/ejh.12246.
- Lee TY, Lai MI, Ramachandran V, et al. Rapid detection of alpha-thalassaemia variants using droplet digital PCR. Int J Lab Hematol. 2016;38:435–443. doi:10.1111/ijlh.12520.
- Shang X, Li Q, Cai R, et al. Molecular characterization and clinical presentation of HKalphaalpha and anti-HKalphaalpha alleles in southern Chinese subjects. Clin Genet. 2013;83:472–476. doi:10.1111/cge.12021.
- Hu L, Shang X, Yi S, et al. Two novel copy number variations involving the alpha-globin gene cluster on chromosome 16 cause thalassemia in two Chinese families. Mol Genet Genomics. 2016;291:1443–1450. doi:10.1007/s00438-016-1193-0.
- Colosimo A, Gatta V, Guida V, et al. Application of MLPA assay to characterize unsolved alpha-globin gene rearrangements. Blood Cells Mol Dis. 2011;46:139–144. doi:10.1016/j.bcmd.2010.11.006.
- Steinberg-Shemer O, Ulirsch JC, Noy-Lotan S, et al. Whole-exome sequencing identifies an alpha-globin cluster triplication resulting in increased clinical severity of beta-thalassemia. Cold Spring Harb Mol Case Stud. 2017;3:a001941, doi:10.1101/mcs.a001941.