
ABSTRACT
Objective
The 3.7 kb deletion (-α3.7) in the α-globin cluster, which characterizes α+-thalassemia, has been reported to have a carrier rate of 4.78% in southern China. Three -α3.7 subtypes have been identified worldwide. However, the -α3.7 III subtype has not previously been identified in China. Herein, we reported identification of the -α3.7 III subtype in a Chinese patient.
Methods
We used gap-PCR and a liquid chip system to detect α-thalassemia mutations. Multiple ligation-dependent probe amplification was performed to detect the large deletion. We finally used Sanger sequencing and single molecule real-time sequencing to characterize and confirm the genotype.
Results
The proband, characterized as -α3.7 III heterozygous, showed microcytosis and hypochromic red cells, with a mean corpuscular volume of 78 fL and mean corpuscular hemoglobin of 25.4 pg. The proband’s mutation was inherited from her father, who had normal blood parameters.
Conclusion
We first identified the -α3.7 III subtype in China. Consequently, all -α3.7 subtypes have now been identified in the Chinese population. Therefore, attention should be paid to -α3.7 III in clinical prenatal diagnosis, given that commonly used methods such as gap-PCR may lead to misdiagnosis or missed diagnosis.
Introduction
The α-thalassemia is among the most common inherited disorders worldwide [Citation1]. It occurs mainly in tropical and subtropical regions, including southern China; a high carrier rate of 11.31% and 17.55%, respectively, has been reported in Guangdong and Guangxi provinces [Citation2,Citation3]. This disorder arises from the deletion or non-deletion in the α-globin cluster, which is located on chromosome 16. The most common α-thalassemia mutation is -α3.7 (5.32%), followed by –SEA (4.82%) and -α4.2 (3.23%) in southern China [Citation4]. On the basis of previous reports, -α3.7 can be divided into three subtypes (-α3.7I, -α3.7II and -α3.7III) [Citation5] depending on the deletion breakpoints. Despite having different junctions, all subtypes result in the same type of α+-thalassemia. Patients with -α3.7 usually present with no clinical manifestations, or light microcytosis and hypochromic red cells. In the Chinese population, -α3.7I is the main subtype, and -α3.7II is rare, whereas no -α3.7III had been reported. However, in the southwest Pacific, where β-globin variants and α0-thalassemia are rare, and malaria is an epidemic, -α3.7III is found at high frequency, for example, 11.5% in Vanuatu [Citation6]. Extensive micro-epidemiological research has shown that α+-thalassemia was likely to have been selected through its interaction with malaria [Citation7]. In Polynesian islands in the central and eastern Pacific Ocean, which are malaria-free, -α3.7III also occurs, with a gene frequency of 7.5% [Citation6]. More than 99% of the α+-thalassemia deletions are of the -α3.7III subtype in French Polynesia [Citation8], which comprises 120 islands spreads over 4000000 km2 in the eastern Pacific Ocean. Vanuatu and Polynesia are presumed to be linked because proto-Polynesian colonizers traveled from Vanuatu en route to the central Pacific, thus potentially explaining the high frequency of the -α3.7III subtype in Polynesia. In South Asia, the -α3.7III subtype has been reported only in the Thai population, with a proportion of approximately 2% in southern Thailand and 0.9% in northeast Thailand [Citation9]. Herein, we report the first identification of the -α3.7III subtype in a Chinese patient by using single molecule real time sequencing (SMRT). We also analyzed the phenotypic effects of this subtype by performing pedigree analysis.
Materials and methods
Patients
The proband was a 24-year-old woman from Huizhou, Guangdong province, China. Genetic analysis to detect the common types of α- (-α3.7, -α4.2, –SEA, αCSα, αQSα and αWSα) and β-(CD41–42 (-CTTT), IVS-II-654 (C > T), -28 (A > G), CD71–72 (+A), CD17 (AAG > TAG), CD26 (GAG > AAG), CD31 (-C), CD27/28 (+C), IVS-I-1 (G > T), CD43 (GAG > TAG), -32 (C > A), -29 (A > G), -30 (T > C), CD14–15 (+G), Cap+40–43 (-AAACA), initiation codon (ATG > AGG) and IVS-I-5 (G > C)) thalassemia revealed normal α- and β-globin genes. However, the hematological parameters detected through routine blood examination indicated hypochromic polyglobulia and microcytic red blood cells (RBCs). In addition, hemoglobin (Hb) quantification indicated slightly low Hb A2 levels. Therefore, the patient came to our hospital for further study after informed consent was obtained according to the Declaration of Helsinki. Her family members were also recruited for pedigree analysis.
Hematological analysis
A peripheral blood sample was collected, and hematological parameters were analyzed with a Sysmex XN5000 automated hematology analyzer (Sysmex Corporation, Kobe, Japan), which has high precision and reproducibility. Hb quantification was performed with an automated capillary electrophoresis system (Sebia Capillarys 2, France).
Molecule analysis
Common α-globin and β-globin mutations were detected with gap-polymerase chain reaction (PCR) and PCR-reverse dot blot (PCR-RDB) with kits from Yaneng Biosciences, China, and a liquid chip system. The primers used for the gap-PCR of detection -α3.7 were as follows, 3.7F (5’-CCCCTCGCCAAGTCCACCC-3’) and 3.7R (5’-AAAGCACTCTAGGGTCCAGCG-3’). Droplet digital PCR [Citation10] was used according to the manufacturer’s instructions to detect rearrangements of the α-globin gene cluster. Sanger sequencing was performed to detect the genotype by using the following primer: F: 5’-TGGAGGGTGGAGACGTCCTG-3’ and R: 5’-TCCATCCCCTCCTCCCGCCCCTGCCTTTTC-3’.
Single molecule real-time sequencing
SMRT was used to confirm the genotype. We first amplified the α-globin gene with PCR (PCR primers were as follow: 3.7III F (5’-CCCAGAGCCAGGTTTGTTTATCTG-3’), 3.7III R (5’-AAAGCACTCTAGGGTCCAGCG-3’)). After amplification and purification, we added a loop adapter to the DNA and prepared the library. Sequencing was performed on a PacBio Sequel System by Berry Genomic Corporation (Beijing, China).
Results
The proband was a 24-year-old woman with hypochromic polyglobulia and microcytic RBCs (mean corpuscular volume (MCV): 78 fL, mean corpuscular hemoglobin (MCH): 25.4 pg). Hb quantification revealed a slightly low Hb A2 level (2.4%). We then performed gap-PCR and PCR-RDB, as well as liquid chip analysis to detect α- and β-thalassemia mutations. No mutations were found with gap-PCR and PCR-RDB ((A)). However, the results the liquid chip analysis indicated that the proband was heterozygous for -α3.7. To further explain the contradictory results obtained through different methods, we performed droplet digital PCR and detected a copy number of 1 for HBA1, thus indicating a heterozygous deletion in HBA1 ((B,C). These findings suggested that the liquid chip results were credible. Therefore, we further performed Sanger sequencing and found a heterozygous fusion between HBA1 and HBA2 ((A)). Finally, to uncover the breakpoint of the deletion, we performed SMRT, a long read, single molecule method. The SMRT analysis recapitulated the droplet digital PCR results, and indicated an approximately 3.7 kb deletion extending from the 3´ untranslated region (3´UTR) of HBA2 to the full-length HBA1 ((B)). The position of the deletion was between positions 173708 and 177519 on chromosome 16 (NG_000006.1:g.34570_38381del, hg38). According to the ithanet database (https://www.ithanet.eu/), the deletion was classified as the third type of -α3.7 (-α3.7III). In addition, we determined that the proband inherited this deletion from her father, who was also diagnosed as heterozygous with -α3.7III, as was her younger brother, whereas her mother was heterozygous for –SEA (). However, her father’s MCV and MCH levels were normal, whereas her younger brother had slightly low MCH levels. The different manifestations among the three people indicated the variation in -α3.7III phenotypes.
Figure 1. Molecular characterization of -α3.7III deletion. (A) Gap-PCR analysis indicating a unique 1826bp product in the proband (XYL), her father (XSN) and her brother (XZT). –SEA/, heterozygous for –SEA; 1826 bp indicates the normal band, and 1306 bp indicates the SEA band. (B, C) Droplet digital PCR results showing the distribution of the positive and negative droplets (B), and the copy numbers of HBA1 and HBA2 in the proband (XYL) and her father (XSN) and brother (XZT). Blue droplets indicate positivity for HBA1 or HBA2, whereas green droplets represent RPP30, the reference gene used to calculate the copy number. Data are shown as mean ± SD.
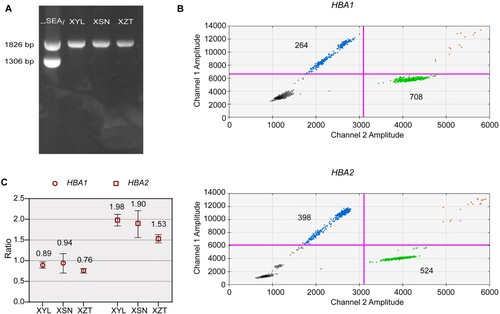
Figure 2. Sequencing analysis of the -α3.7III deletion. (A) Sanger sequencing results of -α3.7III in the proband. (B) SMRT analysis of the patient. The light yellow and blue regions indicate the two alleles of the α-globin gene cluster. The arrows show the region of the deletion. The purple dots indicate sequencing errors. The relative positions of the HBA2 and HBA1 genes on chromosome 16 are indicated by blue boxes. The vertical colored lines indicate nucleotides A (green), T (red), C (blue) and G (orange) discordant with alignment to hg38 reference sequence.
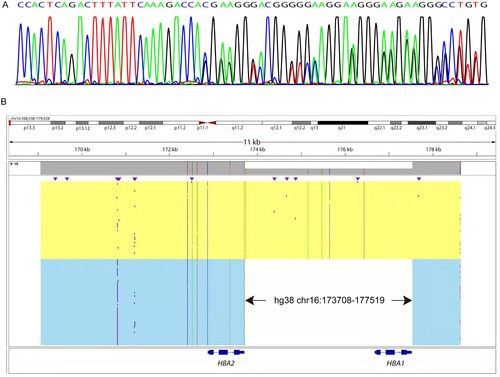
Table 1. The phenotypic and genotypic data of the proband and her families.
Discussion
According to research by Clegg et al. [Citation6], the -α3.7III subtype mainly occurs in Vanuatu and islands of the southwest Pacific Ocean in regions where malaria is endemic [Citation6]. However, the -α3.7III subtype has been reported in the Polynesian islands of the central and eastern Pacific, which are malaria-free, where it accounts for 99% of the α+-thalassemia deletions [Citation8]. A possible explanation for this paradoxical finding is that the early Polynesian colonizers are believed to have traveled from Southeast Asia via the island Melanesia en route to the central Pacific approximately 3,500 years ago. In addition, a previous study has found no example of -α3.7III from various Black, European and Asian individuals [Citation11]. In Southeast Asia, the -α3.7III subtype is rare. These data might indicate that the -α3.7III subtype is confined to Vanuatu and Polynesia.
A common method used to detect -α3.7 is gap-PCR, which performed well in detecting -α3.7I and -α3.7II, but can easily to miss -α3.7III. The primers used to perform PCR were designed to target the third exon of HBA2. Meanwhile, the breakpoint of -α3.7III was located on the 3´UTR of HBA2. We speculated that the position of the primer might have overlap with the breakpoint of -α3.7III and result in a missed diagnosis. In our study, the liquid chip results indicated that the proband, her father and her younger brother were heterozygous for -α3.7, even though gap-PCR indicated normal results. The reason for this finding might have been that the primers used in liquid chip spanned the breakpoint of -α3.7III. Therefore, the liquid chip system was more sensitive than gap-PCR. However, although the liquid chip method was able to detect -α3.7, it was unable to distinguish among the three subtypes. With the rapid development of the single molecule, long read characterization method SMRT [Citation12], the precise breakpoints can now easily be distinguished. The -α3.7III subtype has been reported to be confined to Vanuatu, Polynesia and Thailand. Herein, we identified this type of deletion in a Chinese woman. We also confirmed that the proband and her family were native to Huizhou. Therefore, we believe that some cases with this subtype may be undiagnosed when common methods are used to detect the α-thalassemia deletions. More -α3.7III subtypes may be found in the future with the rapid development of sequencing methods. Our findings indicated that attention should be paid to this subtype of -α3.7 deletion in clinical diagnosis in the Chinese population. Although the -α3.7III heterozygosity behaved with no or light hypochromic polyglobulia microcytic RBC, it turned out to be Hb H disease when compound with –SEA, with which someone may potentially rely on blood transfusion to alive.
In conclusion, we reported the first identification of -α3.7III in the Chinese population by using SMRT. Our findings indicated that SMRT was a useful tool for accurate detection of large deletions. Given the severe clinical features and phenotypes of -α3.7III compounded with –SEA deletions, more insights should be gained during the prenatal period.
Ethics statement
Informed consent was obtained according to the Declaration of Helsinki.
Acknowledgements
The authors thank the patient for his willingness to participate in this study. Financial support from the National Natural Science Foundation of China (grant number 82100136) and the Science and Technology Program of Guangzhou, China (grant number 202002030390) is gratefully acknowledged. We thank International Science Editing for editing this manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
All data in this study are shown in the figures and tables.
Additional information
Funding
References
- Higgs DR, Gibbons RJ. The molecular basis of alpha-thalassemia: a model for understanding human molecular genetics. Hematol Oncol Clin North Am. 2010;24:1033–1054.
- Yin A, Li B, Luo M, et al. The prevalence and molecular spectrum of alpha- and beta-globin gene mutations in 14,332 families of Guangdong Province, China. Plos One. 2014;9:e89855.
- Xiong F, Sun M, Zhang X, et al. Molecular epidemiological survey of haemoglobinopathies in the Guangxi Zhuang autonomous region of southern China. Clin Genet. 2010;78:139–148.
- Shang X, Peng Z, Ye Y, et al. Rapid targeted next-generation sequencing platform for molecular screening and clinical genotyping in subjects with hemoglobinopathies. EBioMedicine. 2017;23:150–159.
- Harteveld CL, Higgs DR. Alpha-thalassaemia. Orphanet J Rare Dis. 2010;5:13.
- Hill AV, Bowden DK, Trent RJ, et al. Melanesians and polynesians share a unique alpha-thalassemia mutation. Am J Hum Genet. 1985;37:571–580.
- Kariuki SN, Williams TN. Human genetics and malaria resistance. Hum Genet. 2020;139:801–811.
- Philippon G, Martinson JJ, Rugless MJ, et al. Alpha-thalassaemia and globin gene rearrangements in French Polynesia. Eur J Haematol. 1995;55:171–177.
- Charoenwijitkul T, Singha K, Fucharoen G, et al. Molecular characteristics of alpha(+)-thalassemia (3.7 kb deletion) in Southeast Asia: Molecular subtypes, haplotypic heterogeneity, multiple founder effects and laboratory diagnostics. Clin Biochem. 2019;71:31–37.
- Bao X, Qin D, Ma J, et al. Accurate detection of alpha-globin gene copy number variants with two reactions using droplet digital PCR. Hematology. 2022;27:198–203.
- Shaulov A, Filon D, Rund D. Haplotype analysis of alpha-thalassemia chromosomes reveals heterogeneity and multiple founders in Ashkenazi Jews. Eur J Med Genet. 2016;59:555–558.
- Xu L, Mao A, Liu H, et al. Long-molecule sequencing: A new approach for identification of clinically significant DNA variants in alpha-thalassemia and beta-thalassemia carriers. J Mol Diagn. 2020;22:1087–1095.