
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
In this work, the known and unknown structural as well as biological properties of 3-(2-aminoethyl) indole (tryptamine) were interpreted using molecular spectroscopy (FT-IR, FT-Raman, NMR and UV–Visible) and cheminformatic tools. The supportive drug-related information was gained by analysing the obtained data which will be useful for the drug chemist for the pharmaceutical research. The important biological properties of the present chemical species satisfied the Lipinski five rules and it was opt to fabricate complex antibiotic compounds. The acquired charge potential load for creating antibiotic strain on compositional parts was keenly observed from the obtained data and it was evaluated by the vibrational analysis and Mulliken charge profile. From the NMR data, the chemical nodal points were noted and their movement around the molecule was carefully monitored. The degenerate and non-degenerate energy profile of orbital interaction system was studied and the link of chemical reactivity path was identified. The significance of excited electronic transitions among non-bonding molecular orbital system was justified and their transitional energy coefficient was determined. The toxicity level was checked from the chirality characteristics and enantiomer structure obtained from vibrational circular dichroism profile.
1. Introduction
The present molecule is fabricated on indole base which has a strong pharmaceutical background [Citation1]. The indole derivatives are popularly used as drug intermediates [Citation2]. In an antimicrobial drug family, they are mostly used in the pharmaceutical industry as starting materials for the drug production [Citation3].
The 3-(2-aminoethyl) indole is fundamentally a biogenic amine, also called as tryptamine, and its structure contains an indole ring along with chains of ethyl groups ended with an amino group. The strong coupling of the amine group with pyrrole produced a rigid chemical potential which demonstrates biological activity which leads to extend the drug’s action. Tryptamine is monoamine alkaloid and essentially behaves as a neurotransmitter also used as a psychedelic drug [Citation4,Citation5]. Though it has 5-hydroxytryptamine receptors’ agonist activity [Citation6], tryptamine is acting as an antitumour agent due to its high cytotoxicity [Citation7,Citation8]. It is also used for the treatment of migraine and obesity with its high pharmacological indices [Citation9].
Though tryptamine is acting as a potential drug due to its series of asymmetrical arrangement of ethyl and amino groups, it has not been properly investigated spectroscopically in order to explore the biological and structural properties using quantum chemical methods. The quantum chemical calculations are usually used for the prediction of known properties which were not explored in the experiment. The computations having enriched calculation methods for finding the physico-chemical parameters and thereby the unknown chemical properties can be studied. In this work, from the obtained results, the related properties were interpreted using experimental and theoretical tools which will be useful for fabricating advanced drug using tryptamine.
2. Experimental profile
2.1. Physical state
The light pink crystal powder form compound, 3-(2-aminoethyl) indole, was purchased and it was pure and of spectroscopic grade.
2.2. Recording details
The FT-IR and FT-Raman spectra of the compound were recorded by making series of scanning in a Bruker-IFS (66 V) spectrometer with different resolution techniques and the instrument by default adopted with FRA 106 Raman module equipped with a Nd:YAG laser source operating at 1.064 µm line widths with 200 mW power [Citation10].
The high-resolution 1H NMR and 13C NMR spectra were captured by 300 and 75 MHz FT-NMR spectrometer [Citation11], respectively.
The UV– Visible spectrum was recorded in the range of 100–800 nm, with the scanning interval of 0.20 nm, using the UV-1700 series instrument [Citation12].
3. Computational profile
Here, the computational details were very important since they were used to furnish all the structure-related parameters for the physical and chemical properties. The molecular structure designed by Gauss View was optimized by Gaussian 09 D. 01.version software program using core i7 computer [Citation13]. Several scanning programs were performed for getting optimized structure and finally the most optimized structure was achieved. The entire structure-related parameters have been calculated and were tabulated for the analysis.
The second-order and third-order molecular orbital interactions were keenly observed for finding the frontier molecular transitions on the electronic structure. The MEP grid view was displayed with multicolour gradient for justifying chemical properties. The computational calculations were performed on the structure by selecting most appropriate methods: DFT; B3LYP and B3PW91 with 6-31++G(d, p) and 6-311++G(d,p) basis sets. The electronic energy profile diagram of the present compound related with absorption spectra and the NBMO energies has been evaluated by the time-dependent SCF method with the best-fit basis set. The chemical energy reaction path mechanism was determined by recording 1H and 13C NMR spectra using the GIAO method adopted with I-PCM model. The effects of hyper- and hypopolarization factors in different coordinates were calculated using the B3LYP method with the 6-311++G(d,p) basis set. The ECD and vibrational circular dichroism (VCD) spectra were simulated from available data of VCD calculations with higher basis set.
The QSAR parameters were calculated by HyperChem (version8.0.6) which facilitated to study several biological properties. The Molinspiration software was also used to accomplish drug-like and evaluation parameters.
4. Results and discussion
4.1. Structural arrangement analysis
In aromatic complex, it is well known that the molecular combinations attained from the addition of suitable substitutions on biologically active molecule can result the novel fusion molecule with improved biological activities. In view of fusing the biological activities of homo–heterocycles and ethyl-amino groups, the combination of such coupling of two molecules may well result in potential novel 3-(2-aminoethyl) indole hybrid derivative with enhanced biological activities. Figure shows the injection of ethyl amino chain on the indole ring in which the impact was clearly seen on plane as well as different coordinates of the molecule. The optimized parameters are displayed in Table and related diagram is shown in Figure .
Figure 1. Molecular structure of 3-(2-aminoethyl) indole.
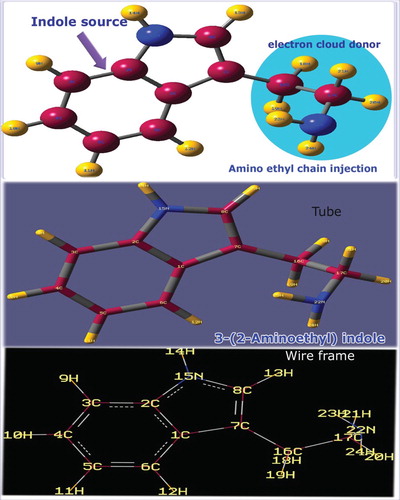
Table 1. Optimized geometrical parameters for 3-(2-aminoethyl) indole computed at HF/DFT (B3LYP&B3PW91) with 6-31++G (d,p) & 6-311++G (d, p) basis sets.
The large bond length was observed in the case of C1–C7 (1.477 Å) on which the chain was substituted, whereas the nearby bond length C7–C8 was 1.372 Å which was found to be very small. The bond length of heteronuclear bond C8–N15 was 1.385 Å and is comparatively high. This increment of bond length was mainly due to the controlling of bond length C7–C8 and it was also clear that the existence of heteronuclear bond drag the stretching of such C–C bond length which may hold consistent dipole moment. The attachment of substitutions on the ring made strong chemical inducement which enables variant chemical atmosphere in the bridge point of ring. This steady arrangement around the fused rings was found to be making strong chemical potential causing cytotoxicity.
The core bond length C–C of hexagonal ring was found to be disturbed much due to the holding of pyrrole ring as well as ligand chain. The impact of the ligand addition in the molecule was observed up to the hexagonal ring. It was evidenced in the bond angle related to the substitutions such as C2–C1–C6, C6–C1–C7, C1–C2–C3, C1–C2–N15, C2–C3–C4 and C1–C6–C5. This semicircle distortion was observed from 107°to 133° in the molecular plane. Though the ethyl groups were disturbed by the amino group, the bond angles among ethyl groups were the same and such as 105° perpendicular to the plane of the molecule. This makes a strong vector form of stress to arrange ethyl groups in opposite coordinate to the amino group. So, two nodal points were made to transfer the chemical potential of amino group to the ring via ethyl groups. In such nodal regions, the supported chemical energy was found to be exchanged and it was supported by the previous study [Citation14]. This energy transformation may be the reason for the inducement of antitumour activity.
4.2. Mulliken charge analysis
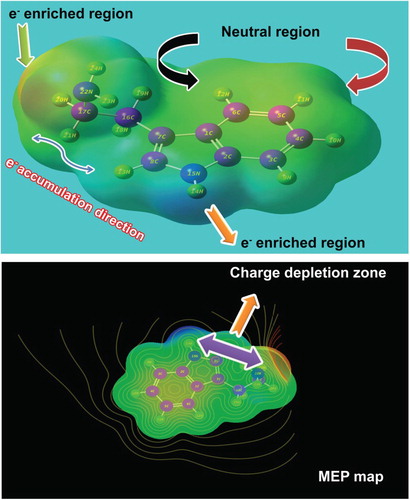
Usually the desired chemical property is persuaded by the oscillating re-configured electronic charges among the molecular sites which are normally measured by the colour gradient generated around the electronic as well as protonic charge zones. Such colour gradient is limited from blue to red colour regions which are represented by Mulliken charge gradient. The colour regions of the present molecule are presented in Figure . Here, the charge dispersion polarization, according to the arrangement of molecular sites, clearly showed the oscillating sequence of Mulliken charge levels in different entities.
Figure 2. Mulliken charge profile diagram of 3-(2-aminoethyl) indole.
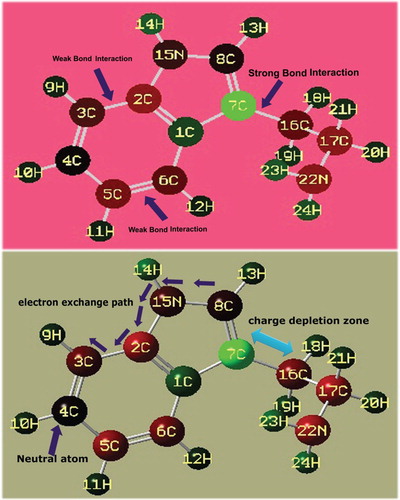
Here, due to the single valence, the hydrogen atoms appeared to be green showing the intensive protonic region. The core C atoms of the ring as well as chain were appeared to be green, red and black and are represented by protonic, electronic and neutral regions, respectively. In the six-member ring, except C1, the C2, C3, C5 and C6 were negative where the electronic content appeared consistently. C4 was a neutral atom upon which the charges were oscillated between the left and right moieties. In the five-member ring, the charges were found to be oscillated from C2, C15 and C8 to the hexagonal ring. Therefore, As the charges oscillated between ring and ligand, charge depletion was noted between ring and ligand. It was clear that the chemical energy was exchanged between chain and ring and the resultant chemical potential emerged from the molecule to be the antibiotic agent.
In the ligand chain, irrespective of C and N, all the core atoms were found to be more negative which means that the chemical energy was accumulated up in nodal points (C and N). This stored energy enabled the chain to be more effective for providing additive chemical potency to the ring for the molecule to be antitumour drug.
4.3. Structure activity/property relationship
The structural parameters related to biological properties were calculated using HyperChem 8.0.6 software, as depicted in Table and the CPK 3D view of the present molecule is displayed in Figure . The Lipinski's rule of five (the Pfizer’s rule) is very important for the drug molecule since it provides the information whether the molecule has pharmacological or biological activity [Citation15]. The RO5 rule expressed the important molecular properties essential for a drug's pharmacokinetics in the human body, together with their absorption, distribution, metabolism, and excretion.
Figure 3. Molecular Lipophilicity Potential of 3-(2-aminoethyl) indole.
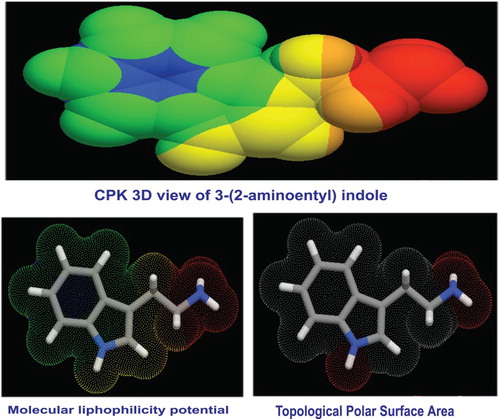
Table 2. Structural parameters of 3-(2-aminoethyl) indole.
According to the rules, the aromatic drug complex is not more than 5-hydrogen bond donors, not more than 10-hydrogen bond acceptors, the molecular mass is less than 500 Dalton, the octanol–water partition coefficient log P is not greater than 5 [Citation16] and the rotatable bond count is not greater than 5. Accordingly, in this case, the hydrogen bond donor count was 1, acceptor was 2, molecular mass was 174.116 Dalton, the log P value was 1.07 and rotatable bond count was 2. The entire parameters related to RO5 were found to be within the expected limit that showed the present compound was orally active drug in humans.
The topological polar surface area (TPSA) of a molecule is a vector sum of the surface of the entire polar atoms; nitrogen and oxygen atoms along with coupling of hydrogen atoms. It is mostly used in pharmaceutical chemistry metric system for measuring the ability to have viable permeability into cells [Citation17]. If the molecule is a successful drug, TPSA should be less than 140 Å2 to penetrate the semi-permeable membrane [Citation18]. In this case, the TPSA was 19 Å2 which is very less and the title molecule is definitely drug. The lipophilicity is an important parameter that should be maintained in passive diffusion in the intestine and the lipophilicity plays an essential role in the administration of kinetic and dynamic aspects of drug action [Citation19,Citation20]. It is a basic property of chemical substance; lipophilicity is a descriptor which measures the transport and impact of chemicals in physiological systems. The lipophilicity is a key factor to determine the absorption and distribution of a drug in the body. The chemical substance penetration across vital membranes and biological barriers, metabolism and excretion is very important for the intake of a drug agent. Here 1.57 is the index sufficient to show the molecule to be a good drug.
The heavy atom count of the drug molecule is a fragmental contribution of structure–receptor interaction that correlates biological activity and the chemical environment. Accordingly, the heavy atom count was 13 which is very high and molecule had potential energy for producing reliable biological activity. The rotatable ligand bond count usually measures the molecular structure complexity that leads to dynamic chirality of the molecule. It directly described the rate of toxicity in the molecule was 2 for the present case. This value was very low and it was clear that the toxicity was much reduced in the compound.
The aromatic chemical species with smaller heavy atom proportions may have high-quality drug-like properties and they can be considered as approved drugs [Citation21]. In this case, the 13 heavy atoms included carbons of the rings as well as ligand groups. Accordingly, the heavy atom count was found to be very high and molecule was having optimized pharmacological activity. The GPCR is a large family of cell surface receptors that respond to a variety of external signals. By these receptors, the sequence of events can be made and through this, the incredible range of bodily functions can be regulated. Here, the GPCR was 0.07 which is sufficient to transfer the signal to the receptor protein.
The Ion channel modulators usually allow evaluating the ability to transport the ion in various cellular and physiological processes. The Ion channel modulator voltage value was 0.22 which was very good chemi-potential energy was able to modulate the signal of Ion channel which leads to move ions across cell membranes. The kinase inhibitor value of the present drug case was 0.10, which was having adequate enzyme inhibition to block the action of protein kinases and therefore it will affect its level of activity and function. The nuclear receptors normally regulate many genes in many tissues; synthetic ligands usually show beneficial therapeutic effects. Major goals in the nuclear receptor field therefore include attaining a better understanding of the mechanisms underlying their actions in specific cell types and ways in which to selectively modulate their activities [Citation22]. Therefore, the nuclear receptor was 1.07 and it efficiently adopts the mechanism to control gene expression for numerous biological progressions.
The nuclear receptors are multifunctional proteins which play key roles in both embryonic development and adult homeostasis which was found to be 3.56 for the title molecule and it was able to transduce signals of their cognate ligands. The protease inhibitor shows antiviral character of the compound and here the same was found to be 1.97 and it was shown that the present compound will be acting as an antiviral drug. In medicinal chemistry and biochemistry, the protease inhibitors are chemical species or complex which is used for inhibition of the function of proteases; perform the same reaction by completely different catalytic mechanisms [Citation23]. As the observed value is 0.48, the present drug has an antiviral character.
4.4. Vibrational profile of molecular system
The vibrational analysis is essentially used for describing the presence of chemical bonds by observing their characteristic region through which the compositional parts of the compound are acknowledged. Here, Table presents the plausible fundamental vibrational blueprint of tryptamine the scanned FT-IR and FT-Raman vibrational wavenumbers of observed and computed spectra (HF and DFT) of which are displayed in Figures and , respectively. The title molecule was tailored by the addition of ethyl-amino chain on the indole ring. The atoms available in the present case were 24 which are found to be undergoing 66 vibrations. According to the rules of group theory, the molecular structure belongs to CS point group due to the presence of multiple planes. The 66 fundamental modes of vibrations were discreted as follows: 24 modes were identified as stretching patterns, 21 modes were represented by in-plane bending vibrations and 21 peaks recognized as out-of-plane bending vibrations.
Figure 4. FT-IR recorded and computed spectra of 3-(2-aminoethyl) indole.
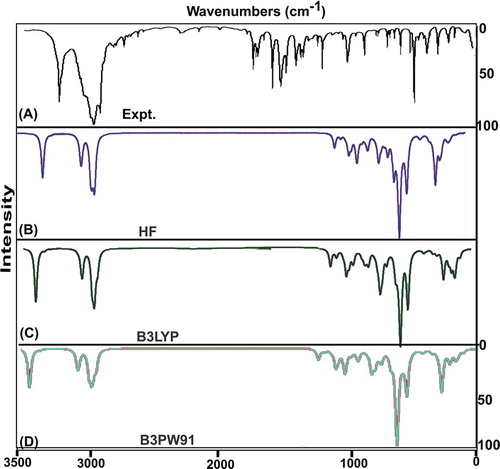
Figure 5. FT-Raman recorded and computed spectra of 3-(2-aminoethyl) indole.
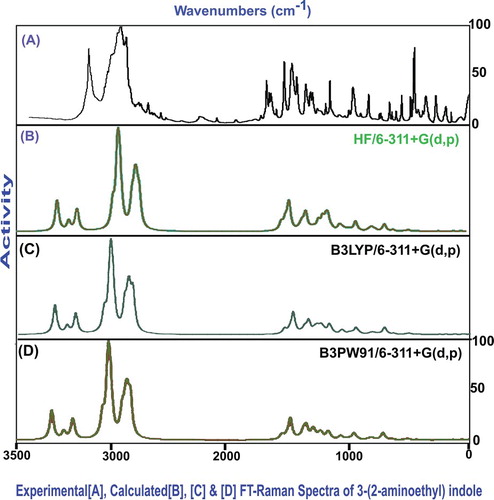
Table 3. Observed and HF and DFT (B3LYP & B3PW91) with 6-31++G(d,p) & 6-311++G(d,p) level calculated vibrational frequencies of 3-(2-aminoethyl) indole.
All the vibrational modes were recognized with respect to their characteristics region which was collected in different regions of IR as well as Raman regions. The entire absorbed and transmitted spectra were found to be under the control of mutual exclusion principle. The entire molecular structure was sketched by the bond length and bond angle vibrations in assigned vibrational region.
4.4.1. N–H and C–N vibrational pattern
The indole and its derivatives usually observed the absorption peaks for N–H stretching mode in the region of 3480–3020 cm−1 [Citation24,Citation25] and their scissoring and wagging modes are usually committed in the region 1460–1320 and 880–710 cm−1, respectively [Citation26]. Here, the N–H bond was present in two different locations (one from pyrrole and another from chain) and thereby the stretching is expected in two distinct places. Accordingly, the first set of N–H stretching vibrations were found at 3300 and 3295 cm−1 and the second N–H vibrations was observed with strong intensity at 3195 cm−1. Though, as expected, these vibrations were identified in two different characteristic regions, both the vibrational modes have been assigned within the predictable limit. As in the literature work [Citation27], this vibrational epic outlook of this molecule indirectly explained that the amine group has taken priority to induce and stabilize the drug character. In addition to that the in-plane (scissoring) and out-of-plane bending (wagging) modes were found with very strong intensity at 1340, 1310 and 1260 cm−1 and 940, 870 and 840 cm−1, respectively. These bending vibrational bands were also uncontaminated by other signals and proved their stability to fascinate new drug property.
Normally, the existence of C–N bond becomes more important in the indole ring since its electronegative polarization as well as it sustained more chemical potential towards amplification of antibiotic activity. In the case of indole, related to C–N vibrations, the strong bands are observed in the spectral region 1280–1180 cm−1 [Citation28] due to conjugation of the electron pair between nitrogen and the core C of the ring. Here, the C–N σ-bond was located in the indole ring and the amino group. Therefore, the C–N stretching signals are observed in different regions of spectrum. Consequently, they were found with very strong to medium intensity at 1605 and 1580 cm−1 and 1460 cm−1. All the bands were occupied in the top end of expected region of spectrum with strong intensity and this state explained the stabilization of chemical reactivity.
4.4.2. C–H vibrations
The present compound is fundamentally indole which is composed of benzene and pyrrole ring. Therefore, the compound has five C–H bonds, four from benzene and one from pyrrole. So, those vibrations are expected to be observed in the region 3100–3000 cm−1 and 3000–2900 cm−1 [Citation29]. The C–H stretching modes were identified at 3035, 2940, 2930, 2925 and 2920 cm−1, respectively. First, four stretching bands were observed at the tail end of expected region and even some of those peaks suppressed much and moved well below the observed region. Usually these vibrations are rather affected; in this case, the substituted chain is also the background reason for the decrement of the wavenumbers.
The indole-linked bending vibrations, such as in-plane and out-of-plane bending vibrations, have been assigned at 1240, 1230, 1150, 1130 and 1120 cm−1 and 850, 820, 810, 770 and 760 cm−1, respectively. These in-plane and out-of-plane bending vibrations are regularly observed in the region 1280 – 1000 cm−1 and 950–700 cm−1, respectively [Citation30]. The entire C–H vibrational frequencies were observed obviously within the expected region and some of those were found at the top end of the allowed range. The entire vibrational bands situated in the mid-range of IR region and the vibrational characteristics of in-plane and out-of-plane oscillations illustrated that the energy associated with vibrations has not consumed by the substitutions attached with the indole.
4.4.3. Core vibrations
Generally, the C = C stretching vibrations in aromatic compounds, particularly, benzene derivatives, are shown in the region 1430–1650 cm−1 [Citation31]. The title compound was benzene adopted pyrrole, in which the benzene core was dominated. So, the core CC stretching vibrations of title compound were observed in both rings. Here, the C = C stretching modes were allotted in the wavenumbers; 1640, 1635, 1610 and 1605 cm−1. Also, the stretching vibrational bands for C–C bond were observed at 1575, 1570, 1500 and 1400 cm−1. Except the last one, the entire bands polarized at the top end of the expected range when compared to the literature values [Citation32,Citation33]. The CCC in-plane bending vibrations are observed at 620, 610, 575 and 570 cm−1 and the out-of-plane bending vibrations are observed at 400, 310 and 260 cm−1. All the in-plane and out-of-plane ring-breathing modes purposively concealed well below the characteristics region since the ring was not able to breathe as usual when it is substituted. Hence, in this case, the potential energy of core ring was exchanged to the chain due to the partial polarization of the forcefully oriented charges.
4.4.4. Ethyl group vibrations
The one and only substitution of the present molecule was ethyl-amino group chain by which the entire assignment of core vibrational frequencies was found to be altered. Here, the ethyl groups were attached directly with the amino group and usually the amino group dominates the ethyl group vibrations [Citation34]. Hence, in this case, the C–H stretching for ethyl groups was observed at 2885, 2845, 2840 and 2820 cm−1 and its connected in-plane and out-of-plane bending vibrations were observed with very strong to strong intensity at 1090, 1020, 1010 and 950 cm−1 and 750, 740, 697 and 670 cm−1, respectively, whereas the stretching, in-plane and out-of-plane bending vibrations have taken the frequency region 2960–2850 cm−1, 1290–910 cm−1 and 900–670 cm−1 , respectively [Citation35]. Correspondingly, the C–C(H2) stretching vibrations were observed at 1380 and 1350 cm−1. The C–CH2 in-plane bending vibrations were observed at 525 and 470 cm−1 and out-of-plane bending vibrations have been observed at 140 and 110 cm−1. These assignments did not agree with the literature [Citation36]. In this case, some of the vibrations were disturbed, which means that the energy related to the C–H and C–C bonds and bond angles was utilized for linking the potential energy of amino group with the ring. From this discussion, it was clear that the potential energy of ethyl group was transducing as a chemical potential to generate drug properties.
4.5. NMR investigation
The observed data from 13C and 1H NMR spectra were used for estimating the chemical environment of chemical compound and also for validating the chemical potential rate of the molecular entities in the large number of chemical and biochemical systems. The nodal point of core atoms can be identified through which the reaction rate mechanism takes place in various locations for resulting drug activities [Citation37]. The chemical potential energy generated in the molecular compositional parts for producing desired chemical property can be decoded by observing Chemical Shift. The chemical reactivity is usually measured gradually from the chemical shifts associated with their positions of the core carbons as well as ligand carbons. The 13C and 1H NMR spectral data were arranged in Table and the corresponding spectral pattern is displayed in Figure .
Figure 6. 13C and 1H NMR spectra of 3-(2-aminoethyl) indole.
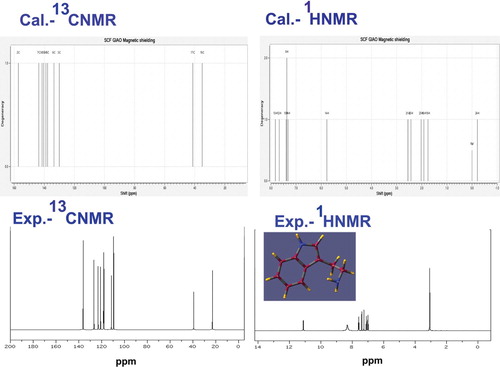
Table 4. Experimental and calculated 1H and 13C NMR chemical shifts (ppm) of 3-(2-Aminoethyl) indole.
Here, the indole ring was fabricated from the fusion of benzene ring with pyrrole ring and the ethyl-amino groups were chained with base. Likewise the chemical system was fabricated for treating tumour cells. The chemical shift of C2 was 157 ppm which is more shifted than other C of the rings. This is the core C of the benzene ring on which the pyrrole ring was substituted through N. This was mainly due to the multiple attractions of charges by the electronically enclosed atoms. This state was also viewed in the case of C8 and thus it showed the dual strong dipole moments (C–N) subsisted in the ring and this high value of force constant amplified the chemical reactivity. The C7 was found to be 143 ppm shifted due to the coupling of chain. The chemical shift of C3, C4, C5 and C6 was observed in the range of 138–130 ppm which is due to the symmetrical attachment of C and H in the ring. The lowest chemical shift of 34 and 41 ppm was observed on C16 and C17, respectively. This lower chemical shift observation was due to the uncertainty of charge accumulations and also these were found to be nodal point of charged region for chemical potential transmission. The chemical energy in the form of signal always transmitted from lower chemical shift nodal point to higher chemical shift nodal region [Citation38]. Accordingly, the source of nodal point was found to be started from C17 and C16 and ended with C2 via C7 of the ring. Among these nodal points, the electronic energy was oscillated and the resultant chemical potential was oriented for inducing antitumour activity.
4.6. Frontier molecular interaction profile
The molecular orbitals are generally named as Frontier molecular orbitals where two sets of characterized orbitals are established and reconfigured, HOMO and LUMO. The LUMO is a set of molecular orbitals where the space is available to hold up the electrons which are excited from the set of highly occupied molecular orbitals (HOMO). The HOMO and LUMO are settled up irrespective of base and ligand groups of the molecule and they are interacted with each other according to the required chemical potential to acquire desired property. The physico-chemical properties (chemical potential) enabling desired drug applications are derived from the equipotential chemical energy associated with the compilation of transitions among electronic sets of orbitals [Citation39]. Both the orbital domains have different finite energy levels on which the electrons are arranged with respect to the available energy scheduled according to the chemical reaction potential. In this part, it is essential to identify the location of HOMO and LUMO over different entities of the molecule, evaluate the chemical kinetic energy associated with the transitions and validate the cause to produce the peculiar drug property. The FMO arrangement for the present molecule is shown in Figure and the computed energy of different orbital levels is represented in Table .
Figure 7. FMO distribution dslay of 3-(2-aminoethyl) indole.
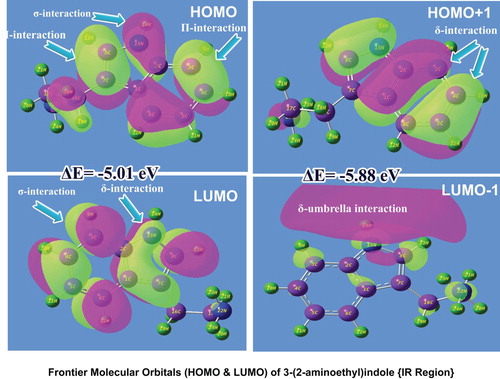
Table 5. Frontier molecular orbitals of 3-(2-aminoethyl) indole with energy levels.
In the present molecular case, the HOMO was configured over the indole ring, where the σ and π-bonding interactions on C=C–C, C–C=C and C–N–C semicircle bonds were identified. In addition to that a small amount of excitation flavour was observed in the ethyl group of ligand. Particularly, σ-bond interaction was taking place on heteronuclear conjugation such as C–N, N–H and C–H, whereas π-bond interaction was observed on C = C only. From these interaction profiles, it was inferred that the required electro-chemical energy was available in HOMO for making the transitions in order to induce the drug property. In the second cascaded HOMO, i.e. HOMO-1, the δ-bonding interaction was recognized on CCC of benzene ring and CCN of pyrrole ring. The δ-bond interaction is very rare to take place on molecular site and this arrangement indirectly indicates new born property.
A number of σ-bond interactions were observed in LUMO which were located on C–C and C–H of the benzene ring, while σ-bond diagonal interactions were found on CCN of the pyrrole ring. It was also noted that the LUMO was absent in the case ethyl-amino groups. In the second-order LUMO, the umbrella-shaped σ and δ-bond interactions were originated. The fundamental interaction energy was found to be started from 5.01 eV to maximum level and this envelope of chemical potential was sufficient to generate the physico-chemical property likely high cytotoxicity. The location of acquired chemical potential was identified to be moved away from the molecule when it is calculated from the first and second order, etc., it was also importantly noted that only part of energy of ligand groups was involved in the property production. The indole ring fully contributed to the typical chemical property such as antitumour activity.
4.7. UV–Visible absorption CT complex profile
The absorption degenerate peaks of electronic excitation in UV–Visible region of title compound are presented in Table and associated experimental and computed CT complex peak pattern are shown in Figure . The entire chemical property of the molecule is the rate of change in the fundamental chemical property of base molecule on par with the ligand groups. In this venture, the charge transfer complex is formed during the doping of ligand with base molecule to make a mechanism for fabricating drug activity [Citation12]. The identification and evaluation of CT complex of chemical compound under study is very vital to examine whether the rate of alternation of fundamental property of the base molecule is changed with the percentage of reaction rate of ligand groups.
Figure 8. UV-Visible spectra of 3-(2-aminoethyl) indole.
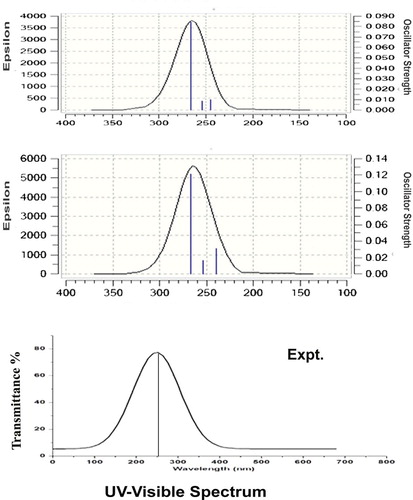
Table 6. Theoretical parameters of electronic absorption spectra of 3-(2-aminoethyl) indole using the TD-DFT/B3LYP/6-311++G(d,p) method.
Here, the present compound was synthesized by interfusing of ethyl-amino chain with the indole ring. Mostly, the CT complex is cultivated on ligand groups since the reaction catalyst is ultimately the substitutional groups. In this case, The electronic absorption signals were acknowledged at 266, 254 and 244 nm with 89%, 87% and 79% molar efficiency with oscillator strength of 0.08, 0.008 and 0.009 on the energy gap of 4.6, 4.8 and 5.0 eV, respectively, the experimental peaks were observed at 255 with 86% molar efficiency and the entire transitions belong to n→σ*. The observed UV–Visible band was nearly equal to the calculated peaks and the band was (R-band-German, radikalartig) assigned to Quartz-UV region of the spectrum. According to the obtained results, the absorption spectra were found to be located in the assigned region of C–N bond and also it was determined that the degenerate peaks confirmed the dual C–N bonds in the compound. From the Mulliken charge profile and the vibrational analysis, the C–N bonds were recognized as CT complex and it was evidenced from the consistent charge accumulation and strong dipole moments. The coherent and unique UV–Visible spectra were observed for gas as well as solvent phase (DMSO and CCl4) and it was proved that the molecule has no effect on solvents.
In addition to that normally, the substitutions should have major impact on molecular properties in the compound which was evidenced from the previous studies [Citation13,Citation40]. But, in this case, the important component C–N of the indole ring would be the CT complex and this component was enhanced by the ligand groups such ethyl-amino groups. From this discussion, it was inferred that the antibiotic property was induced by the significant CT complex which was found to be enriched as antitumour compound by the addition of ligand groups. Owing to the addition of ethyl-amino chain, the oscillated chemical potential was stocked up on the ring itself which was emphasized by the observation of bathochromic shift in the UV–Visible spectra. If the CT band is located in the Quartz-UV region, the compound will react biologically [Citation41]. Therefore, the chemical reactive mechanism constructed by CT complex in the present compound showed cytotoxicity.
4.8. Molecular electrostatic potential maps
The MEP is a space around the electrically charged molecule within which the static electric potential influence can be felt. It is a great tool of colour gradient diagram by which a very useful property can be analysed for predicting molecular reactive behaviour. The electrostatic potential has been used particularly for the measurement of reaction rate of molecule with protein complex and also it is a gauge for estimating colour temperature gradient of electrophilic and nucleophilic regions. It is very much useful for evaluating the intermolecular interactions between drug and its cellular receptor [Citation42]. The molecular electrostatic potential map and its spreading of field composite molecule were displayed in between ambiguity positive and predominated negative regions in Figure . The potential colour gradient can be drafted using the following equation in the computational calculations.
In the figure, the protonic content was located at the N–H bond of the indole which was due to the vacancy of the electron cloud and appeared blue whereas the intensive electrophilic region was found over the amino group of chain. In the case of chain, the electron intensity was retained at amino group which was found to be protected by the ethyl group. In this point of chain, the charge gradient was depleted against the benzene ring of indole. The electrophilic region was always located on amino group if present [Citation43], where the region is directly associated with the nitrogen lone pair. The intermediate neutral region was found on CC of the indole ring and chain which is the indication of the basic nature of the core covalent carbons. The entire hydrogen bonds of molecule also appeared as the intermediate region. This interaction was established by the σ-interacting system of the compound which was measured to be ±6.58 Joules. In addition to that the negative and positive potential was dispersed between N–H and N–H of the pyrrole ring and the substituted amino group, respectively, which makes static dipole moment of 1.79 dyne. For the successful drug chemicals, the static dipole moment of more than unity is enough for the Drug–receptor and enzyme–substrate interaction process. So the present compound has good affinity for the biological systems.
Figure 9. MEP display of 3-(2-aminoethyl) indole.
4.9. Physico-chemical properties
Usually, the molecular system was constructed with the separation of two sets of energy domains on which the entire electronic energy levels are reconfigured after the formation of product compound. The entire electronic transitions take place around these two energy domains, HOMO and LUMO. The set of higher energy levels (HOMO) is related to the more reactive molecule in the reactions with electrophiles, whereas the lower energy profile (LUMO) is essential for molecular reactions with nucleophiles [Citation44]. The transitions between these two profiles are used to measure the physico-chemical properties of the chemical compound. The calculated parameters in IR and UV–Visible regions are presented in Table .
Table 7. Physico-chemical parameters of 3-(2-Aminoethyl) indole.
The zero point vibrational energy of the present compound in IR and UV–Visible region was −497.32 and −497.00 kcal/mol, respectively. From these observed values, it was emphasized that the present structure of the compound was optimized without Cis and Trans. Structures. The dipole moment of the molecule was calculated by the DFT method, 1.59 and 1.79 dyne in IR and UV– Visible region, respectively. The dipole moment is correctly used for measuring the charge separation with respect to the centre of symmetry. The computed values illustrated that the charge depletion takes place to enhance molecule stability and thus, it showed the consistent chemical rigidity-enriched biological activity. The chemical hardness is one of the highly useful parameters which make it possible for the chemists to recognize chemical resistance of the chemical compound. Here, it was 2.50 and 2.63 eV in IR and UV–Visible region, respectively. The measured value in different regions of the present compound proved that the reluctance to react with added chemical species and values showed the coherent chemical stability in both regions.
The force constant of chemical bonds is used to evaluate the chemical reactive energy of intermolecular interactions which can be calculated by the ionization potential. The present case was 0.91 and 0.95. These values are comparatively low, but as the present molecule has a low molecular volume, the observed values are theoretically to be sufficient to sustain the gained chemical reactive energy. The electronegativity of a molecule is an imperative parameter which assesses the asymmetrical orientation of electron cloud with respect to electro-chemical equilibrium forces which is mainly used for the prediction of electro-chemical rigidness of molecule [Citation45]. Here, this was found to be 3.22 and 3.58 in IR and UV region, respectively. These computed values were found to be very much high and were able to react with related protein complex. It also confirmed the impulsive orientation of electron gradient to fabricate noted drug activity. This was achieved by the existence of couple of σ-bond (C–N) interaction in the indole ring and is well known form MEP diagram.
The electrophilicity index is an effective parameter which is mainly used to measure biological toxicity and also as a potential descriptor for toxicity prediction [Citation44]. Accordingly, in this case, the electrophilicity index is 0.82 and 0.92 eV in IR and UV–Visible region, respectively. In both regions, the present compound was having low index value which showed less toxicity. It was also evidenced that in this case, the low toxicity was evaluated from the number of rotatable bonds and number of heavy atoms.
The chemical kinetic potential energy transformation is clearly evidenced from the electrophilicity charge transfer which was +3.34 and 2.49 in IR and UV–Visible region, respectively, for the present molecule. From this value, it was ensured that substantial amount of chemical kinetic was swapped from ethyl-amino group chain to the indole ring and it was for persuading the antitumour activity.
4.10. Polarization and hyper polarization analysis
On the application of radiation energy, the electric components are arranged symmetrically and anti-symmetrically in the molecular sites in terms of existence of electro-chemical mechanical forces generated by the electric strain. Such a process is freezing the first- and second-order sequence multi-pole moments. The first order is called polarizability, whereas the second order is called hyperpolarizability. The first-order polarization is usually guiding to accumulate chemical potential among the active molecular sites, whereas the hyperpolarization is transducing the negative chemical potential into action potential. Thus the measurement of both parameters is used to estimate the molecular properties and biological activities [Citation46].
The average polarizability and anisotropic-polarizability of the present compound were determined using standard methods in computational calculations which were 165 × 10−33 esu and 237 × 10−33 esu, respectively, and the hyperpolarizability (β) was 283 ×10−33 esu. Usually, the first-order polarizability is always smaller than the second-order polarizability since the energy needed for the transduction is lesser than the energy required for producing the action potential [Citation47]. In this case, the average polarizability was computed with respect to entire atoms in the molecule. But the anisotropic-polarizability is effectively calculated from the asymmetrical charge gradient. Here, though it was a simple molecule, both the polarizabilities were so high and make hypoaction mechanism which induces crude biological properties. The higher order polarizability with large coefficient generating hyperactive mechanism in the molecular sites strongly enhanced antibiotic components in the molecule. This was accomplished by ethyl-amino groups in the indole ring. From this discussion, it was concluded that since the compound has hyper asymmetrical polarization coefficient, it was strongly emphasized in the novel pharmaceutical property (Table ).
Table 8. The dipole moments µ (D), the Polarizability α(a.u.), the average Polarizability αo (esu), the anisotropy of the Polarizability Δα (esu), and the first hyperpolarizability β(esu) of 3-(2-Aminoethyl) indole.
4.11. NBMO transition analysis
The structural property is associated with physico-chemical properties which are composed of the electronic transitions among the important energy levels: Lewis donor and non-Lewis acceptor orbitals such that the transitions are frequently taking place among important chemical bonds in the molecules called non-bonding molecular orbitals. This analysis is basically used to observe in which entities making root cause of the drug potential. The important exchange of chemical potentials in NBMO profile was listed along with the interaction energies in Table .
Table 9. The calculated NBMO of 3-(2-Aminoethyl) indole by the second-order Perturbation theory.
In this case, several transitions were observed within the ring system as well as between ring and chain of the present compound. In the first case, within core CC of the ring, the energies of 10.4, 6.3 and 11.9 kcal/mol were transferred from C1–C2 to C3–C4, C5–C6 and C7–C8, respectively, which were assigned to σ–σ* transition in σ-interaction system. Similarly, the energy of 3.25 and 2.95 kcal/mol was found to be transferred from C3–C4 to C1–C2 and C5–C6 and was assigned as σ–σ *. Another transitions have been observed from C5–C6 to C1–C2 and C3–C4 in which 10.5 and 10.2 kcal/mol energy were respectively used for π–π * interaction system. The important transitions were observed from C17–N22 to H24 and C16–C17 by consuming 103.3 and 19.6 kcal/mol amount of energy between two rings. In the same way, the transitions were found from C17–N22 to N22–H23 and N22–H24 by uptaking energy 19.4 and 67.4 kcal/mol. When compared all the above transitions, the maximum amount of energy was transferred from NH2 end to CH end chain.
The transitions from N22–H24 to H24 were observed in reverse order by absorbing energy 14.4 and 102.5 kcal/mol. Similarly, the transitions were observed from C5–N6 to N22–H23 and N22–H24 by absorbing 21.3, 17.9 and 69.6 kcal/mol amount of energy and assigned to σ–σ*. The second-order transitions from N22 to lone pair of H23 and H24 by consuming 48.3 and 387.4 kcal/mol. In this part, the maximum amount of energy per mol was found to be observed and this energy impact may be generated by intensive chemical strain for property establishment. It was also evidenced from other transitions among lone pair of N22 to N22–H23, N22–H24, C1–C2 and C7–C8 using the energy of 39.2, 424.2, 13.49 and 13.42 kcal/mol. These transitions also appeared in the counterpart. In this case, a large amount of transitional energy was observed within the indole ring particularly in the benzene wing, ethyl –amino group predominantly within the amino group. This view clearly showed the important activation nodes by which the entire antitumour property was stimulated.
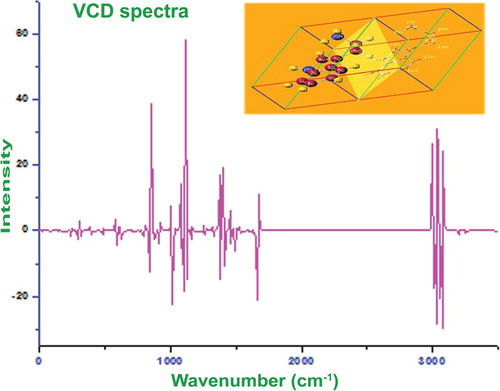
4.12. Vibrational circular dichroism analysis
The aptitude of VCD to provide a delegate structural signature formulates it an influential tool in modern biochemistry and pharmaceutical chemistry and its applications are universally found in every field of study. In the VCD observation, the structural elements and their role of activity in the drug property are precisely distinguished since their recorded emission and absorption bands do not overlap widely at whole wavenumbers. It is also used to estimate virtual and real drug toxicity.
The calculated regular VCD spectrum of optimized structure is exhibited in Figure . Each and every optimized structure has its own specific VCD signature and it does not change its signal pattern unless otherwise any complex bonded with the molecular system. Mostly, the reduction of complexity in chiral system is a complicated process and thereby it is very difficult to identify the optimized structure to terminate drug toxicity. But, in this case, the molecule was found to be very simple and belong to two planes only. The VCD sequence was not too complicate and it was very easy to optimize the structure. Accordingly, the enantiomer view was investigated in terms of vibrational sequence. In the VCD figure, the substitutional region appeared to be clear and in chronological order and the emission and absorption peaks were observed with same coefficient. In the fingerprint region, though there were few dissimilar crests found and they appeared to be distorted, when it was seen the vibrational frequency as a whole, the molecule had quite helix. This manifestation does not produce any unprincipled toxic nature and this compound can be used to fabricate good drug with chemical purity.
Figure 10. VCD spectrum of 3-(2-aminoethyl) indole.
5. Conclusion
The compound under investigation is basically a drug compound particularly it is antibiotic compound. It was properly analysed in order to explore the entire structural and biological properties. From the structural and biological parameters, the entire physico-chemical and biological properties were interpreted and the properties were evaluated by performing different analysis using spectroscopic and chemic-quantum computational study. The active compositional parts of the compound were identified from energetic presence of the chemical bonds in their characteristic regions of the spectrum. The intermolecular interaction profile was keenly observed and the orbital overlapping process for the first- and second-order frontier molecular system was studied. Accordingly, the residing of chemical energy was determined for the cause of drug potential. The chemical nodes in the molecular site were recognized by observing the NMR data. The fruitful transitions in the non-bonding molecular orbitals were acknowledged and the exchange of chemi-kinetic potential for regulating and stabilizing drug property was noted. The hiding toxicity was estimated and it was very low in this present compound and it was concluded that the 3-(2-aminoethyl) indole (tryptamine) was opt for fabricating drug derivatives.
Disclosure statement
No potential conflict of interest was reported by the authors.
ORCID
K. Hemachandran http://orcid.org/0000-0002-7699-1563
P. Anbusrinivasan http://orcid.org/0000-0001-6109-6729
C. Manoharan http://orcid.org/0000-0002-5671-872X
References
- Lee J-H, Lee J. Indole as an intercellular signal in microbial communities. FEMS Microbiol Rev. 2010;34(4):426–444. doi: 10.1111/j.1574-6976.2009.00204.x
- Parke DV. The biochemistry of foreign compounds. Oxford: Pergamon Press; 1968, p. 45–52.
- Nelson L, David M, Cox M. Principles of biochemistry. 4th ed. New York: W. H. Freeman; 2005.
- Jones RS. Tryptamine: a neuromodulator or neurotransmitter in mammalian brain? Prog Neurobiol. 1982;19(1–2):117–139. doi: 10.1016/0301-0082(82)90023-5
- Pauwels PJ, John GW. Clin Neuropharmacol. 1999;22:123–132.
- Adams D, Enardeau AB, Bickerdike MJ, et al. 5-HT2C receptor Agonists for the treatment of obesity. biological and chemical Adventures. Chim. Int. J. Chem. 2004;58:613–620. doi: 10.2533/000942904777677506
- Reyes F, Martin R, Fern R. Granulatamides A and B, cytotoxic tryptamine derivatives from the soft Coral Eunicellagranulata. J Nat Prod 2006;69:668–670. doi: 10.1021/np050382s
- Beck B, Hess S, Domling A. One-pot synthesis and biological evaluation of aspergillamides and analogues. Bioorg Med Chem Lett 2000;10:1701–1705. doi: 10.1016/S0960-894X(00)00305-X
- Adla SK, Sasse F, Kelter G, et al. Doubly prenylated tryptamines: cytotoxicity, antimicrobial activity and cyclisation to the marine natural product flustramine A. Org Biomol Chem 2013;11:6119. doi: 10.1039/c3ob40896e
- Aarthi R, Ramalingam S, Periandy S. Acta Sci Pharma Sci. 2018;2(1):13–23.
- Manzoorali M, George G, Ramalingam S, et al. Spectroscopic investigation and chemical properties analysis on anticancer compound; α,α,ά,ά-Tetrabromo-p-Xylene with computational analysis. J Mol Struct. 2016;1106:37–52. doi: 10.1016/j.molstruc.2015.10.078
- Susithra G, Ramalingam S, Periandy S, et al. J Pharmacol Med Chem. 2018;2(1):1–17.
- Madanagopal A, Periandy S, Gayathri P, et al. Molecular structure activity on pharmaceutical applications of Phenacetin using spectroscopic investigation. J Mol Struct. 2017;1127:611–625. doi: 10.1016/j.molstruc.2016.08.028
- Karthikeyan N, Joseph Prince J, Ramalingam S, et al. Electronic [UV–visible] and vibrational [FT-IR, FT-Raman] investigation and NMR–mass spectroscopic analysis of terephthalic acid using quantum Gaussian calculations. Spectrochim Acta, Part A. 2015;139:229–242. doi: 10.1016/j.saa.2014.11.112
- Lipinski CA, Lombardo F, Dominy BW, et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 2001;46(1-3):3–26. doi: 10.1016/S0169-409X(00)00129-0
- Leo A, Hansch C, Elkins D. Partition coefficients and their uses. Chem Rev. 1971;71(6):525–616. doi: 10.1021/cr60274a001
- Pajouhesh H, Lenz GR. Medicinal chemical properties of successful central nervous system drugs. NeuroRx. 2005;2(4):541–553. doi: 10.1602/neurorx.2.4.541
- Hitchcock SA, Pennington LD. Structure−brain exposure relationships. J Med Chem 2006;49(26):7559–7583. doi: 10.1021/jm060642i
- Lombardo F, Gifford E, Shalaeva MY. In silico ADME prediction: data, models, facts and myths. Mini Rev Med Chem. 2003;3:861–875. doi: 10.2174/1389557033487629
- Taylor NR, Smith R. The World Wide Web as a graphical user interface to program macros for molecular graphics, molecular modeling, and structure-based drug design. J Mol Graph. 1996;14:291–296. doi: 10.1016/S0263-7855(96)00077-X
- Mao F, Ni W, Xu X, et al. Molecules. 2016;21(75):1–18.
- Burris TP, Busby SA, Griffin PR. Targeting orphan nuclear receptors for treatment of metabolic diseases and autoimmunity. Chem Biol. 2012;19:51–59. doi: 10.1016/j.chembiol.2011.12.011
- Oda K. New families of carboxyl peptidases: serine-carboxyl peptidases and glutamic peptidases. J Biochem. 2012;151(1):13–25. doi: 10.1093/jb/mvr129
- Millich F, Becker EI. Synthesis and infrared spectra of some indole compounds1. J Org Chem 1958;23:1096–1102. doi: 10.1021/jo01102a003
- Katritzky AR, Ambler AP. Physical methods in heterocyclic chemistry. New York: Academic Press; 1963, p. 161.
- George Socrates. Infrared and Raman characteristic group frequencies (tables and charts). London- New York: John Wiley & Sons, Ltd; 2001.
- Johnson P, George G, Ramalingam S, et al. J Mol Pharm Org Process Res. 2018;6(1):1–17.
- Pouchert CJ. The Aldrich library of FTIR spectra. New Zealand: The Aldrich Co.; 1985.
- Xue Y, Xu D, Xie D, et al. Density functional theory studies on tautomeric stability and infrared spectra of 2-chloroadenine. Spectrochim Acta A. 2000;56:1929–1938. doi: 10.1016/S1386-1425(00)00252-3
- Xue Y, Xie D, Yan G. Density functional theory studies on molecular structure and IR spectra of 9-methyladenine: a scaled quantum mechanical force field approach. Int J Quantum Chem 2000;76:686–699. doi: 10.1002/(SICI)1097-461X(2000)76:6<686::AID-QUA2>3.0.CO;2-B
- Krishna kumar V, John Xavier R. Indian J Pure Appl Phys. 2003;41:95–99.
- Sathyanarayana DN. Vibrational spectroscopy theory and applications. 2nd ed. New Delhi: New AgeInternational (P) limited publisher; 2004.
- Kalsi PS. Spectroscopy of organic compounds. New Delhi: Wiley Eastern Limited; 1993, 117–118.
- Roeges NGP. A guide to the complete interpretation of the infrared spectra of organic structures. New York: Wiley; 1994.
- Green JHS, Harrison DJ, Kynoston W. Vibrational spectra of benzene derivatives—XII 1,2,4-trisubstituted compounds. Spectrochim Acta. 1971;27A:807–815. doi: 10.1016/0584-8539(71)80159-9
- Shanmugam R, Sathayanarayana D. Spectrochimica Acta A. 1984;40–49.
- Kameytani T, Kajiwara M, Takahashi T, et al. Relationship between reactivity and 13C chemical shifts of benzocyclobutenes. Tetrahedron. 1975;31:949–951. doi: 10.1016/0040-4020(75)80106-2
- Johnson P, George G, Ramalingam S, et al. J Pharm Pharm Res. 2018;1-10001:1–12.
- Fleming I. Frontier orbitals and Organic chemical reactions. London- New York: John Wiley & Sons; 1976.
- Madanagopal A, Periandy S, Gayathri P, et al. Spectroscopic and computational investigation of the structure and pharmacological activity of 1-benzylimidazole. J Taibah Univ Sci. 2017;11:975–996. doi: 10.1016/j.jtusci.2017.02.006
- Manzoor Ali M, George G, Ramalingam S, et al. Vibrational [FT-IR, FT-Raman] analysis, NMR and mass – spectroscopic investigation on 3,6-dimethylphenanthrene using computational calculation. J Mol Struct. 2015;1099:463–481. doi: 10.1016/j.molstruc.2015.05.066
- Bentley J. Determination of electronic energies from experimental electron densities. J Chem Phys 1979;70:159–164. doi: 10.1063/1.437216
- Politzer P, Laurence PR, Jayasuriya K. Molecular electrostatic potentials: an effective tool for the elucidation of biochemical phenomena. Environ Health Perspect. 1985;61:191–202. doi: 10.1289/ehp.8561191
- Rauk A. Orbital interaction theory of Organic chemistry. 2nd ed. New York: John Wiley & Sons; 2001, p. 34.
- Shalini A, Tandon H, Chakraborty T. J Bioequiv Availab. 2017;9(6):536–546.
- Wang J, Xie XQ, Hou T, et al. Fast Approaches for molecular polarizability calculations. Fast J. Phys. Chem. A. 2007;111:4443–4448. doi: 10.1021/jp068423w
- Moorthy N, Jobe Prabakar PC, Ramalingam S, et al. J Theor Comput Sci. 2015;2(4):1–13.