
ABSTRACT
Objectives
To compare the pharmacokinetic and safety of the test group capecitabine tablets (0.5 g) and the reference group capecitabine tablets (0.5 g).
Methods
This study was registered at www.chinadrugtrials.org.cn under the registration number CTR20220138. 48 subjects with solid tumor were recruited and randomized to receive either the test group or the reference group at a dose of 2 g per cycle for three cycles of the entire trial.
Results
The point estimate of the geometric mean ratio of Cmax for the subject and reference groups was 1.0670, which was in the range of 80.00%-125.00%. And the upper limit of 95% confidence interval was −0.0450 < 0. The statistics of geometric mean ratio of AUC0-t and AUC0-∞ (test group/reference group) and their 90% confidence intervals were in the range of 80.00%-125.00%, thus the test group was bioequivalent to the reference group under the conditions of this postprandial test. There were no major or serious adverse events. Conclusion: The pharmacokinetic profiles of capecitabine under postprandial conditions were consistent between the two groups. The two groups were bioequivalent and had a similar favorable safety profile in Chinese patients with solid tumor.
1. Introduction
Cancer is a public health problem of worldwide concern, and the incidence of cancer is increasing year by year [Citation1], with colorectal cancer (CRC) being the third most common cause of cancer-related deaths [Citation2]. Gastric cancer (GC) follows as the fourth most common cause of cancer-related deaths [Citation2]. Meanwhile, and breast cancer (BC) is the fifth most common cause of cancer-related deaths and the most common malignant tumor worldwide [Citation3,Citation4]. Despite the decline in cancer incidence with advances in medical care, the global burden of malignant tumors is projected to increase through 2040 [Citation4].
Capecitabine (trade name: Xeloda®) is an oral fluorouracil carbamate agent, which is a precursor drug of fluorouracil that is converted to 5-Fluorouracil (5-FU) in tumor tissues by the action of higher concentrations of thymidine phosphorylase in tumor cells [Citation5,Citation6]. In clinical practice, capecitabine is used as a first-line treatment for breast, colorectal and gastric cancers, etc [Citation7,Citation8]. Oral administration of Xeloda® mimics continuous intravenous infusion while avoiding the barrier problems associated with parenteral gastrointestinal administration [Citation9,Citation10]. The molecular structure of the carbamate agent of Xeloda® is rapidly absorbed in the intestinal tract and its bioavailability exceeds 70%. Capecitabine, used in this study, has been shown to treat solid tumor in previous studies. This trial was conducted under postprandial conditions in accordance with FDA and was designed to compare the pharmacokinetics and safety of the test product capecitabine (0.5 g), with the reference product, Xeloda® (0.5 g) in Chinese patients with solid tumor.
2. Materials and methods
2.1. Study design
This was a multicentre, randomized, open, two-agent, single-dose, three-cycle, partially repeated crossover design human bioequivalence study. The study was approved by the Ethics Committee of the First Affiliated Hospital of Bengbu Medical College, Anhui Jimin Cancer Hospital Chenzhou First People’s Hospital Liaoning University of Traditional Chinese Medicine. The protocol of this clinical study was in accordance with the Declaration of Helsinki and the International Conference on Harmonisation of Good Clinical Practice guidelines. Informed consent was obtained from all clinical trial participants before enrollment. Referring to the Qilu Pharmaceutical Co., Ltd. has been approved information publicity is shown in , 48 cases of postprandial 3 × 3 design to obtain the intra-individual coefficient of variation of 33.45%: according to Jiangsu Hengrui Pharmaceutical Co., Ltd. has been approved information publicity, 76 cases of postprandial 2 × 2 design to obtain the intra-individual coefficient of variation of 36.4%; according to the intra-individual coefficient of variation of 35%, 40.0% calculation of the cases as shown in , and finally decided to 48 subjects were recruited for the formal trial.
Table 1. NMPA registration publication statistics.
Table 2. Sample size calculation.
Inclusion criteria: patients over 18 years of age with histopathologically confirmed solid tumor, including colorectal cancer, breast cancer, or other solid tumor for which capecitabine tablet chemotherapy regimens are applicable, and patients may be in a tumor-bearing or non-tumor-bearing status; women weighing no less than 45 kg and men weighing no less than 50 kg; body mass index of 19.0 to 24.0 kg/m2; ECOG score of 0–1; patients with liver metastases: ALT ≤ 5×ULN, AST ≤ 5×ULN, DBil ≤ 3×ULN; renal function: Cr ≤ 1.5×ULN, Ccr >50 ml/min; bone marrow function: white blood cell count ≥ 3.0 × 109/L, neutrophil count ≥ 1.5 × 109/L, platelets ≥ 100 × 109/L, hemoglobin ≥90 g/L. Subjects were excluded if they had: 4) experienced anti-tumor therapy other than capecitabine (including chemotherapy, radiotherapy, molecular targeted therapy, biotherapy, major surgical therapy, immunotherapy, administration of traditional Chinese medicines with anti-tumor effects, etc.) within 2 weeks prior to the first dose, or 1 weeks of capecitabine; known hypersensitivity to fluorouracil or to 5-fluorouracil; complete deficiency of dihydropyrimidine dehydrogenase (DPD) activity; severe cardiovascular disease; presence of brain metastases or other central nervous system metastases; use of any medication that inhibits or induces the hepatic metabolism of the drug within 28 days prior to the screening; folinic acid, interferon-alpha, phenytoin within 2 weeks prior to the first dose, warfarin, coumarins, antacids containing aluminum hydroxide or magnesium hydroxide.
2.2. Study procedures
During screening, each subject received a unique screening number. Data Management and Statistical Analysis Unit generation was dynamically randomized using a central randomization system, allowing each subject to be randomly assigned to the group. A standard meal was consumed 30 min before the next morning’s dose and the meal was finished within 30 min. Thirty min after the start of the meal timer, 4 of the test group (T) or 4 tablets of the reference group (R) were administered in 240 mL of water, allowing for a deviation of ±1 min. The study drug must be swallowed whole and not chewed, crushed or separated. Staff will be required to check the subject’s mouth and container after administration to ensure that all of the medication has been taken.
Blood samples were collected at the time of predose and 0.25, 0.50, 0.75, 1.00, 1.33, 1.67, 2.00, 2.33, 2.67, 3.00, 3.50, 4.00, 5.00, 6.00, 7.00, and 8.00 hour after dose, each point of blood collection about 3 mL. Centrifuge conditions: 2500 g for 10 min at 4°C. Plasma concentrations of capecitabine were determined by LC-MS/MS.
The sample pretreatment procedure for the determination of capecitabine concentration in human plasma by LC-MS/MS was as follows: Plasma samples were allowed to come to room temperature and vortexed to mix. Add 50 μL of sample to the wells of a 96-well plate; for double-blank or ULOQ samples, add 50 μL of 50% methanol solution, and for other samples, add 50 μL of internal standard working solution (the internal standard working solution is 1 μg/mL capecitabine standard), vortex to mix well, then add 300 μL of methanol to each well of the sample, seal the plate, and mix well for 10 min; 2623 g for 10 min at 4°C, take 100 μL of supernatant after centrifugation into another 96-well collection plate, add 200 μL of 50% methanol, seal the plate, mix for 10 min and centrifugation at 2623 g for 10 min at 4°C, and then inject the sample. The linear range of the assay was 20.000 ng/mL to 20,000.000 ng/mL, and the LLOQ was 20.000 ng/mL. The liquid chromatograph is a Thermo Scientific Dionex Ultimate 3000 UHPLC. The mass detector is: AB API 4000, the ion source is: Turbo Spray. Detailed assay procedures are described in .
Table 3. Liquid chromatography analysis of capecitabine.
Table 4. Mass spectrometry analysis of capecitabine.
Vital signs were monitored at 1.0 ± 0.5 h, 4.0 ± 0.5 h, and 8.0 ± 0.5 h after administration.
2.3. Analysis
Pharmacokinetic parameters (PK): the main pharmacokinetic parameters of capecitabine in plasma were calculated using the non-atrial model Phoenix WinNonlin 8.1 (Pharsight, Inc., U.S.A.) software or above: Cmax,T1/2,Tmax,AUC0-t,AUC0-∞,λz.
Safety: record adverse events (AEs): laboratory tests, vital signs, 12-lead ECG results, local tolerance and physical examination and summaries with appropriate descriptive statistics.
3. Results
3.1. Baseline characteristics
In this study, a total of 88 subjects were screened for the postprandial drug administration trial. After a thorough medical examination, 40 subjects failed screening according to the above inclusion and exclusion criteria, of which, 37 subjects failed because they did not meet the inclusion criteria and 3 subjects failed because they voluntarily withdrew. Forty-eight volunteer subjects with solid tumor were finally enrolled (). One subject in the T-R-R group voluntarily withdrew from the trial before the second cycle of administration, and no drugs were administered and no blood was collected in the second and third cycles. Therefore, this subject was excluded from the BES for cycles two and three. The demographic and baseline characteristics of the 48 subjects are shown in .
Figure 1. Flow Diagram of the study.
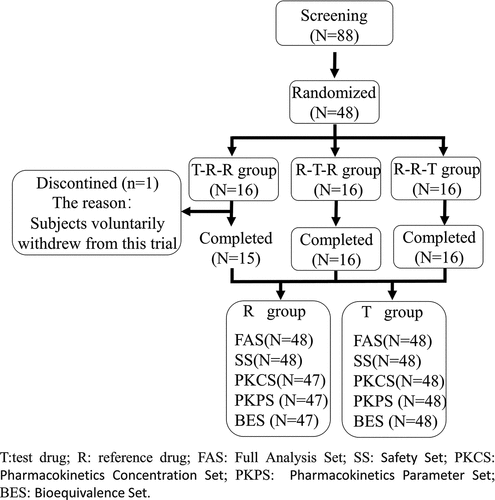
Table 5. Baseline characteristics.
3.2. Pharmacokinetics
In this clinical trial, the plasma drug concentration of capecitabine was determined by LC-MS/MS in 48 subjects after postprandial single-dose oral administration of the test group capecitabine and 47 subjects after posttraumatic single-dose oral administration of the reference group capecitabine, and the bioequivalence of capecitabine in T and R were calculated and analyzed by using WinNonLin 8.1.T and R were administered separately after the administration of, the mean drug concentration-time curves of capecitabine were similar in both groups (, ).
Figure 2. Capecitabine mean drug concentration-time curve in the test drug and reference drug.
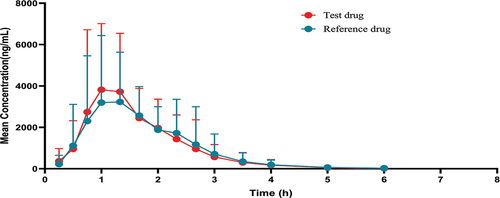
Table 6. The inferential analysis of capecitabine pharmacokinetic parameters of the test group and the reference group (data set: PKS).
The intra-individual standard deviation of capecitabine Cmax of the reference group was 0.311, SWR >0.294, and the point value of the ratio of geometric means of T and R was 1.0670, which was within the range of 80.00%-125.00% and the upper limit of the 95% CI was −0.0450 < 0 by the RSABE method of bioequivalence evaluation; therefore, the conclusion of bioequivalence of Cmax was established. Therefore, the conclusion of bioequivalence of Cmax was established. The intra-individual standard deviation of the reference group AUC0-t was 0.118, SWR <0.294, and the 90% CI of the geometric mean ratio of T and R was 99.62–107.60%, which is within the range of 80.00%-125.00%, so the conclusion of bioequivalence was established for AUC0-t by using the ABE method for bioequivalence evaluation. The intra-individual standard deviation of AUC0-∞ of capecitabine reference group was 0.118, SWR <0.294, and the bioequivalence evaluation was performed by ABE method, and the 90% CI of the geometric mean ratio of T and R was 99.57%-107.62%, which was within the range of 80.00%-125.00%, so the conclusion of bioequivalence of AUC0-∞ was established.
In summary, in this postprandial trial, the rate (Cmax) and extent (AUC0-t and AUC0-∞) of absorption of capecitabine was bioequivalent after a single oral dose of 4 tablets of the test group and a single oral dose of 4 tablets of reference group.
3.3. Safety
During the trial period, 41 AEs occurred in 22 subjects, with an adverse event rate of 45.83% (), one subject in the test group had one sub-grade 3 AEs of upper respiratory tract infection, but the investigator believed that the adverse reaction might not be related to the drug, the rest of the AEs were of grade 2 or less in severity, no serious AEs, severe adverse reactions, and no AEs leading to withdrawal of adverse reactions, AEs, and a good clinical safety profile for subjects after oral administration of T and R under postprandial dosing conditions.
Table 7. Summary of adverse events.
Common AEs in the two groups included lower white blood cell counts, lower central granulocytes, lower blood pressure, and anemia, which are AEs that occur with most chemotherapeutic agents. Although laboratory tests reflected adverse reactions, subjects 41 AEs ended in recovery/loss of visit.
4. Discussion
Capecitabine is relatively non-cytotoxic in vitro, and in vivo the drug is converted to 5-FU by enzymes. Compared with intravenous 5-FU, oral capecitabine is better than intravenous 5-FU in terms of safety and efficacy [Citation11], at the same time, oral capecitabine causes fewer adverse reactions and most of them are reversible, patients with poor liver function can also be taken orally, the more common adverse reaction is hand-foot syndrome, oral vitamin B6 can alleviate the symptom, and panthenol is effective in preventing hand-foot syndrome. There are adverse reactions such as neutropenia, nausea, vomiting and diarrhea in the hematological system, which can be alleviated by reducing the dose of oral capecitabine. Therefore, this experiment was conducted by oral administration.
According to the Xeloda® prescription, monotherapy was 1250 mg/m2 orally twice daily for 2 weeks. It was found that the different doses of capecitabine taken were not expected to have a significant effect on pharmacokinetics [Citation12]. Therefore, in this study, capecitabine was used at a dose of 2000 mg that did not vary with BSA or body weight. Also, referring to the Xeloda® instruction manual pharmacokinetic parameters for capecitabine: T1/2 about 0.75 h, postprandial Tmax delay about 1.5 h [Citation13–15]. A minimum elution period of 5 half-lives is required according to FDA and EMA guidelines and 7 half-lives according to NMPA guidelines, so a 1-day elution period was adequate in this trial.
The t1/2 of capecitabine was similar to that of the early PK study of Xeloda®. ANOVA revealed no significant difference in t1/2 values between the two agents, indicating comparable rates of drug elimination in vivo. Wilcoxon paired test on the raw data showed no statistically significant difference in Tmax between the subject and reference reagents. The mean plasma concentration-time curves were similar after administration of the test product or reference product, and the 90% CI for Cmax, AUC0-t, and AUC0-∞ fell within the bioequivalence acceptable range of 80.00%-125.00%, allowing the two groups of agents to be judged to be bioequivalence.
Based on safety considerations, capecitabine is associated with the potential for adverse reactions both in monotherapy (adjuvant treatment of CRC, treatment of metastatic colorectal cancer and metastatic breast cancer) and in combination chemotherapy regimens for different indications [Citation16,Citation17]. Data from clinical trials analyzed that capecitabine adverse reactions involve multiple systems throughout the body, mainly including metabolic and nutritional abnormalities, neurological abnormalities, gastrointestinal disturbances, hepatobiliary abnormalities, ocular lesions, skin and subcutaneous tissue syndromes, and systemic and site-of-drug-administration abnormalities [Citation18–21]. Therefore, patients with solid tumor are more suitable for this study than healthy subjects.
In this trial, the three most common AEs for the test drug were upper respiratory tract infection, increased and decreased blood pressure. For the reference drug the five most common AEs were decreased white blood cell count, increased blood triglycerides, decreased blood pressure, decreased neutrophil count and upper respiratory tract infection. However, in previous trials, the most common side effects were diarrhea, dehydration, DPD deficiency, skin reactions and hyperbilirubinaemia [Citation22–24]. Possible reasons for such differences in side effects are differences in the high-level structure, biological activity and purity of the test drugs. Alternatively, these differences may be related to the subject population, dietary conditions and sample size. No SAEs occurred during the postprandial trial and no subjects withdrew from the clinical trial due to AEs. The results of the study suggest that postprandial administration of capecitabine in the test product and reference product dosage forms has a good safety profile in Chinese subjects with solid tumor.
Considering the potential racial differences, this study supplemented the PK parameters as well as AEs of Xeloda® in Chinese patients with solid tumor, which provides some reference for clinical guidance the use of drug administration. However, this trial also has some limitations because the number of subjects was limited, so the occurrence of some AEs such as hyperalgesia and supraventricular extrasystole contraction cannot be excluded as small probability events occurring randomly, and the assessment of the relative risk of these side effects occurring in capecitabine subjected subjects should be accompanied by a long-term follow-up study of the subjects. Classification studies of patients with different types of solid tumor could be performed in future trials, enabling a better understanding of pharmacokinetic parameters and adverse effects in patients with different types of tumor.
5. Conclusion
The study statistically analyzed the PK parameters and safety of postprandial oral capecitabine tablets versus Xeloda® in Chinese patients with solid tumor. At the same time, the bioequivalence experimental design was optimized by comparing relevant clinical trials that have been registered and completed. It is hoped that this manuscript will provide a reference for the research and clinical use of capecitabine.
Article highlights
The test group was bioequivalent to the reference formulation by measuring total capecitabine in Chinese patients with breast, colorectal or gastric cancer under postprandial conditions.
Safety profiles and tolerability of the test formulation of capecitabine were adequate.
Summarised the key points for the design of trials for bioequivalence studies of capecitabine.
Abbreviations
| CRC | = | colorectal cancer |
| BC | = | breast cancer |
| GC | = | gastric cancer |
| 5-FU | = | 5-Fluorouracil |
| ECOG | = | eastern cooperative oncology group |
| ALT | = | alanine aminotransferase |
| AST | = | aspartate aminotransferase |
| DBil | = | direct bilirubin |
| Cr | = | creatinine |
| Ccr | = | creatinine clearance |
| DPD | = | dihydropyrimidine dehydrogenase |
| CI | = | Confidence interval |
| T | = | test group |
| R | = | reference group |
| PK | = | pharmacokinetic parameters |
| Cmax | = | maximum concentration |
| t1/2 | = | elimination half-life |
| Tmax | = | time to reach maximum concentration |
| AUC0-t | = | area under the plasma concentration-time curve from time 0 to last time of quantifiable concentration |
| AUC0-∞ | = | area under the plasma concentration-time curve from time 0 extrapolated to infinite time |
| λz | = | elimination rate constant |
| AEs | = | adverse events |
| BSA | = | body surface area |
Declaration of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Authorship contribution statement
HT Li, RF Shan and JX Ding contributed equally. HT Li contributed to data collection, conducted the analysis, and wrote the report. RF Shan managed the drug and biological samples. JX Ding participated in quality control throughout the trial. JL Zhang, Q Ge, H Li and FX Li contributed to study organization and implementation. BY Liu took care of patients and obtained samples. DM Cheng, XL Li, CY Zhang, H Su, XY Li, HR Li and JJ Ye provided clinical support and medical supervision throughout the trial. All authors approve the final version to be published and agree to be accountable for all aspects of the work. Besides, all authors declare no competing interests. H Zhou and Q Huo design the study. Y Su reviewed and revised this manuscript.
Reviewer disclosures
Peer reviewers on this manuscript have received an honorarium from Expert Opinion on Drug Metabolism and Toxicology for their review work but have no other relevant financial relationships to disclose.
Additional information
Funding
References
- Clegg LX, Feuer EJ, Midthune DN, et al. Impact of reporting delay and reporting error on cancer incidence rates and trends. J National Cancer Inst. 2002;94(20):1537–1545. doi: 10.1093/jnci/94.20.1537
- Tao X, Li T, Gandomkar Z, et al. Incidence, mortality, survival, and disease burden of breast cancer in China compared to other developed countries. Asia Pac J Clin Oncol. 2023;19(6):645–654. doi: 10.1111/ajco.13958
- Thrift AP, Wenker TN, El-Serag HB. Global burden of gastric cancer: epidemiological trends, risk factors, screening and prevention. Nat Rev Clin Oncol. 2023;20(5):338–349. doi: 10.1038/s41571-023-00747-0
- Yang L, Kartsonaki C, Yao P, et al. The relative and attributable risks of cardia and non-cardia gastric cancer associated with Helicobacter pylori infection in China: a case-cohort study. Lancet Public Health. 2021;6(12):e888–e896. doi: 10.1016/S2468-2667(21)00164-X
- Teperikidis E, Boulmpou A, Charalampidis P, et al. 5-fluorouracil, capecitabine and vasospasm: a scoping review of pathogenesis, management options and future research considerations. Acta Cardiol. 2022;77(1):1–13. doi: 10.1080/00015385.2021.1873548
- Gieschke R, Reigner B, Blesch KS, et al. Population pharmacokinetic analysis of the major metabolites of capecitabine. J Pharmacokinet Pharmacodyn. 2002;29(1):25–47. doi: 10.1023/A:1015716617967
- Osterlund P, Kinos S, Pfeiffer P, et al. Continuation of fluoropyrimidine treatment with S-1 after cardiotoxicity on capecitabine- or 5-fluorouracil-based therapy in patients with solid tumours: a multicentre retrospective observational cohort study. ESMO Open. 2022;7(3):100427. doi: 10.1016/j.esmoop.2022.100427
- Cassidy J. Potential of Xeloda in colorectal cancer and other solid tumors. Oncology. 1999;57(Suppl 1):27–32. doi: 10.1159/000055266
- Marsé H, Van Cutsem E, Grothey A, et al. Management of adverse events and other practical considerations in patients receiving capecitabine (Xeloda). Eur J Oncol Nurs. 2004;8(Suppl 1):S16–30. doi: 10.1016/j.ejon.2004.06.006
- Sternberg CN, Reichardt P, Holland M. Development of and clinical experience with capecitabine (Xeloda) in the treatment of solid tumours. Eur J Oncol Nurs. 2004;8(Suppl 1):S4–15. doi: 10.1016/j.ejon.2004.06.005
- Van Cutsem E, Hoff PM, Harper P, et al. Oral capecitabine vs intravenous 5-fluorouracil and leucovorin: integrated efficacy data and novel analyses from two large, randomised, phase III trials. Br J Cancer. 2004;90(6):1190–1197. doi: 10.1038/sj.bjc.6601676
- Queckenberg C, Erlinghagen V, Baken BC, et al. Pharmacokinetics and pharmacogenetics of capecitabine and its metabolites following replicate administration of two 500 mg tablet formulations. Cancer Chemother Pharmacol. 2015;76(5):1081–1091. doi: 10.1007/s00280-015-2840-6
- Cassidy J, Twelves C, Cameron D, et al. Bioequivalence of two tablet formulations of capecitabine and exploration of age, gender, body surface area, and creatinine clearance as factors influencing systemic exposure in cancer patients. Cancer Chemother Pharmacol. 1999;44(6):453–460. doi: 10.1007/s002800051118
- Reigner B, Blesch K, Weidekamm E. Clinical pharmacokinetics of capecitabine. Clin Pharmacokinet. 2001;40(2):85–104. doi: 10.2165/00003088-200140020-00002
- Jacobs BAW, Deenen MJ, Joerger M, et al. Pharmacokinetics of capecitabine and four metabolites in a heterogeneous population of cancer patients: a comprehensive analysis. CPT: Pharmacometrics Systems Pharmacology. 2019;8(12):940–950. doi: 10.1002/psp4.12474
- Gambill BD. First-line capecitabine is as effective as 5-fluorouracil/leucovorin in treating advanced colorectal cancer. Clin Colorectal Cancer. 2001;1(1):18–19. doi: 10.1016/S1533-0028(11)70533-3
- Daniele G, Gallo M, Piccirillo MC, et al. Pharmacokinetic evaluation of capecitabine in breast cancer. Expert Opin Drug Metab Toxicol. 2013;9(2):225–235. doi: 10.1517/17425255.2013.759939
- Huang MD, Fuss H, Lademann J, et al. Detection of capecitabine (Xeloda®) on the skin surface after oral administration. J Biomed Opt. 2016;21(4):47002. doi: 10.1117/1.JBO.21.4.047002
- Yaghobi Joybari A, Azadeh P, Ghiasi HA, et al. Capecitabine induced fingerprint changes. J Clin Pharm Therapeutics. 2019;44(5):780–787. doi: 10.1111/jcpt.13003
- Brazelton A, Yande S, Pope R, et al. Racial and ethnic differences in capecitabine toxicity in patients with gastrointestinal tract cancers. Ann Gastroenterol. 2022;35(2):182–186. doi: 10.20524/aog.2022.0688
- Dooley M, Goa KL. Capecitabine. Drugs. 1999;58(1):69–76. discussion 77-68. doi: 10.2165/00003495-199958010-00006
- Nishida M. Pharmacological and clinical properties of Xeloda (capecitabine), a new oral active derivative of fluoropyrimidine. Nihon Yakurigaku Zasshi. 2003;122(6):549–553. doi: 10.1254/fpj.122.549
- Saif MW, Katirtzoglou NA, Syrigos KN. Capecitabine: an overview of the side effects and their management. Anticancer Drugs. 2008;19(5):447–464. doi: 10.1097/CAD.0b013e3282f945aa
- Kadoyama K, Miki I, Tamura T, et al. Adverse event profiles of 5-fluorouracil and capecitabine: data mining of the public version of the FDA adverse event reporting system, AERS, and reproducibility of clinical observations. Int J Med Sci. 2012;9(1):33–39. doi: 10.7150/ijms.9.33