
ABSTRACT
Introduction: DNA double-strand breaks (DSBs) are toxic DNA lesions that can be repaired by non-homologous end-joining (NHEJ) or homologous recombination (HR). Mutations in HR genes elicit a predisposition to cancer; yet, they also result in increased sensitivity to certain DNA damaging agents and poly (ADP-ribose) polymerase (PARP) inhibitors. To optimally implement PARP inhibitor treatment, it is important that patients with HR-deficient tumors are adequately selected.
Areas covered: Herein, the authors describe the HR pathway mechanistically and review the treatment of HR-deficient cancers, with a specific focus on PARP inhibition for BRCA1/2-mutated breast and ovarian cancer. In addition, mechanisms of acquired PARP inhibitor resistance are discussed. Furthermore, combination therapies with PARP inhibitors are reviewed, in the context of both HR-deficient and HR-proficient tumors and methods for proper patient selection are also discussed.
Expert opinion: Currently, only patients with germline or somatic BRCA1/2 mutations are eligible for PARP inhibitor treatment and only a proportion of patients respond. Patients with HR-deficient tumors caused by other (epi)genetic events may also benefit from PARP inhibitor treatment. Ideally, selection of eligible patients for PARP inhibitor treatment include a functional HR read-out, in which cancer cells are interrogated for their ability to perform HR repair and maintain replication fork stability.
1. Introduction
DNA continuously encounters multiple different DNA lesions from endogenous sources (e.g. radical species as byproducts from cellular metabolism) as well as exogenous sources (e.g. ultraviolet radiation and pharmaceutical agents). To preserve genomic stability, cells are equipped with a tightly regulated signaling network that detects and repairs DNA lesions, collectively called the ‘DNA damage response’ (DDR) [Citation1]. To facilitate DNA repair, the DDR activates cell cycle checkpoints to arrest ongoing cell cycle progression. Furthermore, if the number of DNA lesions exceeds the amount that can be managed by the DDR, cells will be cleared from the proliferative compartment by programmed cell death through apoptosis or induction of senescence.
The response to DNA damage is not a linear pathway, and its activation does not lead to fixed phenotypic outcomes. Rather, the DDR consists of multiple parallel pathways which display extensive feedback and cross talk. DDR signaling has widespread influence on cellular homeostasis, as underscored by the observation that the upstream DDR kinases ATM and ATR phosphorylate >700 substrates in various pathways in response to DNA damage [Citation2]. Conversely, DDR pathways receive input from multiple cellular cues, including pro-survival and pro-death signals, which ultimately influence cell fate decisions to promote cell survival or cell death in response to DNA damage.
Genetic defects in DNA repair pathway components or cell cycle checkpoints are associated with a range of clinical phenotypes, including neurodegeneration and cancer predisposition [Citation1]. These observations illustrate the relevance and complexity of genome maintenance pathways. Interestingly, research over the last decades has demonstrated that cancer-associated DNA repair defects not only lie at the basis of tumor development but also give rise to vulnerabilities that can be exploited therapeutically.
1.1. Induction of DNA double-strand breaks
DNA double-strand breaks (DSBs) are potentially highly toxic DNA lesions. DSBs can arise as a consequence of multiple mechanisms. First, DSBs are induced under physiological circumstances during maturation of B- and T-cells during V(D)J recombination, the mechanism that randomly assembles DNA segments to generate diversity in immunoglobulins and T-cell receptors [Citation3]. Specifically, RAG-1 and RAG-2 introduce DSBs that are randomly joined together to shuffle genomic areas and create sequence variation [Citation4]. Second, DSBs arise non-physiologically. Most aberrant DSBs appear to be associated with replication. These breaks can result from unrepaired DNA single-strand breaks (SSBs) that are converted into DSBs during replication. Alternatively, nucleotide depletion, interstrand DNA cross-links, or collisions between the replication and transcription machinery may stall replication forks, which as a result thereof can collapse and lead to single-ended DSBs [Citation5]. Notably, many anticancer therapeutics, including platinum-containing agents and topoisomerase inhibitors, exert their cytostatic effects through interfering with DNA replication and thus cause DSBs. Of note, other anticancer treatments (e.g. irradiation or chemotherapeutic agents such as bleomycin) also cause DSBs in nonreplicating cells, by directly assaulting DNA.
1.2. Repair of DNA DSBs
Repair of DSBs is governed by two fundamentally different pathways: non-homologous end-joining (NHEJ) and homologous recombination (HR). DSBs are repaired by either of these pathways, and the choice between these types of DSB repair depends largely on the cell cycle phase, although additional factors such as chromatin context appear to play a role [Citation6].
1.2.1. Non-homologous end-joining
Classical NHEJ is a very efficient DNA repair pathway that acts throughout the cell cycle and directly ligates DNA ends [Citation7]. NHEJ is present in both eukaryotes and prokaryotes and operates through a largely conserved pathway [Citation8]. In mammalian cells, most DSBs are repaired by NHEJ, since this repair type is active throughout interphase. An important characteristic of NHEJ is that it can ligate breaks with different chemical ends. In the process of NHEJ, DSBs are recognized and bound by Ku70–Ku80 heterodimers, which activate the DNA–PKcs kinase (), right panel). Subsequently, the XRCC4:DNA ligase-IV complex is recruited, together with nucleases and polymerases, to complete DNA-end joining [Citation9]. NHEJ works in a sequence-independent fashion and, since DNA ends may have been damaged and require processing prior to ligation, NHEJ is error-prone and can induce mutations [Citation10]. In contrast to classical NHEJ, alternative NHEJ (alt-NHEJ) involves different players and creates deletions at the repair junction [Citation11].
Figure 1. a) Schematic representation of double-strand break repair and protection of stalled replication forks. Left panel: For repair of DSBs by NHEJ, breaks are recognized and bound by Ku70-Ku80 heterodimers which activate DNA-PKcs. XRCC4, DNA ligase-IV and polymerases (µ/λ) are recruited to complete DNA-end joining. Middle panel: During HR repair, DSBs are recognized by the MRN complex, which initiates DNA-end resection in conjunction with CtIP and BRCA1. EXO1 and DNA2 generate extensive ssDNA stretches, which are coated with RPA. In a PALB2-dependent fashion, BRCA2 is recruited, which loads RAD51 onto the ssDNA to invade the sister chromatid and to find sequence homology. Right panel: In response to stalled replication forks, BRCA1, BRCA2, FANCD2 and RAD51 protect nascent DNA for MRE11-dependent degradation. b) Cell cycle-dependent switch between HR and NHEJ. HR only occurs in S and G2 phases of the cell cycle. The switch between HR and NHEJ depends on the activity of S-phase CDKs, which phosphorylate CtIP to activate the MRN complex and stimulate DNA-end resection. DNA-end resection is negatively regulated by 53BP1 and RIF1, which thereby promote NHEJ. Other cell cycle kinases also control HR, including Plk1 and CK2 which control RAD51 recruitment.
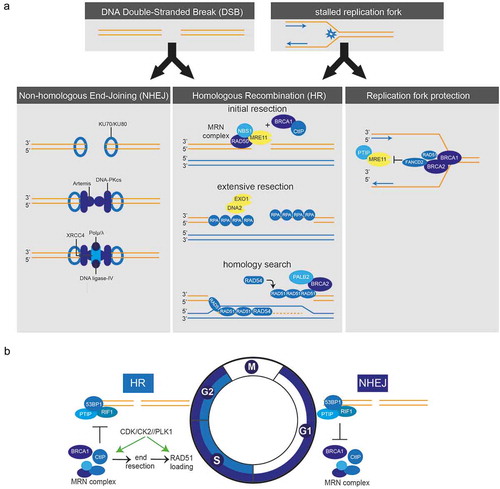
1.2.2. Homologous recombination
In contrast to NHEJ, HR uses a DNA template to repair DSBs, for which the sister chromatid is usually employed. The use of a template makes HR conservative when it comes to DNA sequence and remarkably error-free when compared to NHEJ [Citation12]. Of note, single-strand annealing, an independent DNA repair pathway, also requires extensive homology but results in annealing of homologous single-strand DNA ends, which induces deletions [Citation13]. The requirement of a template restricts HR to S and G2 phases of the cell cycle when DNA replication has occurred ()) [Citation14]. Although a genome-wide template for HR becomes available upon DNA replication, only a subset of DSBs is actually repaired by HR in S/G2. The mechanisms that underlie the usage of HR versus NHEJ in S/G2 cells remain largely unclear, although chromatin composition appears to influence the choice of repair type [Citation6]. In contrast, repair of replication fork-associated DSBs is completely dependent on HR, since these DSBs are single-ended and therefore require template-mediated resolution.
HR is a complex pathway and involves many components (), middle panel). DSBs are recognized by the MRN complex, which consists of MRE11, RAD50, and NBS1. The MRN complex tethers DNA ends and promotes activation and recruitment of ATM to sites of DSBs. Reciprocally, ATM phosphorylates and activates all members of the MRN complex [Citation15,Citation16]. A critical step in the commitment to repair DSB through HR is the formation of ssDNA overhangs at the sites of DNA ends. This process, called DNA-end resection, is initiated by the MRN complex in conjunction with CtIP and BRCA1 [Citation17] ()). MRE11, as part of the MRN complex, has endonuclease activity and can initiate DNA-end resection in 5ʹ to 3ʹ direction and starts ∼200–300 nucleotides away from the DSB site [Citation18]. In doing so, the MRN complex creates relatively short ssDNA overhang at DSB sites, which function as an entry site for the EXO1 and DNA2 helicase/exonuclease enzymes that generate extensive ssDNA stretches [Citation19,Citation20]. Following end resection, the ssDNA is coated with replication protein A (RPA) protein complexes to stabilize ssDNA structures. In parallel, BRCA2 is recruited in a BRCA1- and PALB2-dependent fashion to ultimately recruit RAD51 to the ssDNA overhangs. RAD51 replaces RPA and forms nucleoprotein filaments on the ssDNA, which will invade the homolog sister chromatid to search for sequence homology and initiate strand exchange [Citation21]. Multiple additional factors are involved in controlling HR. For instance, five paralogs of RAD51 exist (i.e. RAD51B, RAD51C, RAD51D, XRCC2, and XRCC3) that appear to support HR. All RAD51 paralogs are essential genes, as deletion of these genes in mice results in embryonic lethality [Citation22]. The recruitment of RAD51 to DSBs is dependent on RAD51 paralogs as well as on RAD52, deficiency of which aggravates the phenotype of BRCA1, BRCA2, or PALB2 deletion [Citation23]. In this context, the RAD51C paralog appears to play the most prominent role. Mechanistically, it was shown to delay progression of the cell cycle during DNA damage by promoting CHK2 phosphorylation during initiation of DDR signaling [Citation24]. Conversely, the HR component RAD54, a protein of the SWI2/SNF2 complex, has ATPase activity which requires the presence of dsDNA [Citation25]. RAD54 interacts with RAD51 to stabilize RAD51 filaments and is involved in strand invasion and, eventually, formation of Holliday junctions [Citation26].
As described above, the loading of RAD51 onto ssDNA is a key step in completing DSB repair by HR. It has been shown that TOPBP1, in conjunction with PLK1, is required for phosphorylation and loading of RAD51. In line with these findings, an siRNA screen identified TOPBP1 as being synthetically lethal with olaparib, which was explained by impaired RAD51 foci formation upon TOPBP1 depletion [Citation27].
Cells that are deficient in HR, for example, due to loss of BRCA1/2, are dependent on alternative pathways to repair DSBs. This includes classical or alternative NHEJ. Indeed, error-prone NHEJ was shown to generate increased genomic instability when HR is defective [Citation28]. The alt-NHEJ pathway requires DNA polymerase θ (Polθ), which prevents RAD51 loading onto ssDNA [Citation29]. When compared to other NHEJ polymerases, Polθ was shown to preferably bind a 5ʹ-terminal phosphate and use the opposite overhang to anneal DNA strands and therefore produces highly mutagenic DNA junctions [Citation30].
1.3. Balancing between HR and NHEJ
DNA-end resection is a point-of-no-return and marks the ultimate decision to repair DSBs through HR ()). This switch is governed in large part by cell cycle-dependent phosphorylation of CtIP by cyclin-dependent kinases (CDKs), which promotes endonuclease activity of MRE11 within the MRN complex to initiate DNA-end resection [Citation17,Citation31]. CtIP is predominantly recruited to DSBs during S and G2, in complex with BRCA1 [Citation32]. Since the activity of CDKs increases when DNA recombination commences, this mechanism ensures restriction of DNA end-resection to cell cycle phases where template DNA is available. The switch between HR and NHEJ is also regulated by additional mechanisms. Specifically, DNA-end resection is negatively regulated by 53BP1 and RIF1, which are both substrates of ATM. RIF1 binds to 53BP1 and ultimately promotes NHEJ [Citation33]. 53BP1 interferes with BRCA1 function and thereby prevents DNA-end resection whereas, conversely, BRCA1 promotes dephosphorylation of 53BP1 to stimulate DNA-end resection [Citation34]. In the recent years, multiple other factors have been identified that regulate DNA-end resection and thereby control HR initiation and poly (ADP-ribose) polymerase (PARP) inhibitor sensitivity. For example, REV7 is recruited to sites of DSBs in a 53BP1-dependent fashion and blocks DNA-end resection [Citation35,Citation36]. Also, the DNA helicase HELB and the demethylase JMJD1C affect chromatin responses to DNA breaks and ensuing DNA-end resection and thereby control RAD51 recruitment to sites of DNA breaks [Citation37,Citation38]. Finally, 53BP1 recruitment was shown to be regulated by ring finger protein 168 (RNF168), an altered abundance of which induced toxic NHEJ, genomic instability, and differential sensitivity towards PARP inhibitors [Citation39]. Exactly how the opposing effects of CtIP/BRCA1 and 53BP1/RIF1/REV7/HELB/JMJD1C operate at the molecular level remains incompletely clear. It has been shown, however, that the repositioning of 53BP1 and end-resection activity depend on ubiquitin ligase activity of BRCA1 together with BARD1 and the subsequent chromatin remodeling by SMARCAD1 [Citation40].
Also other cell cycle regulators have been shown to impact on DSB repair. For example, RAD51 is phosphorylated by polo-like kinase-1 (Plk1) and casein kinase-2 (CK2), which is followed by binding to the MRN component NBS1, which facilitate recruitment to DNA breaks [Citation41]. Although not all molecular mechanisms have been elucidated and novel regulators will likely be identified, it is becoming increasingly clear that the switch between HR and NHEJ not only requires CDK activity but involves multiple stimulatory and inhibitory factors of DNA-end resection and homology search.
1.4. Replication fork stability
Independent of their role in repair of DSBs, HR proteins such as BRCA2 and RAD51 paralogs are involved in the protection of stalled replication forks, thereby preventing chromosomal instability (), right panel). BRCA2, as well as BRCA1 and FANCD2, prevents degradation of nascent DNA at stalled replication forks by stabilizing RAD51 filaments. This pathway is independent of the role of BRCA1/2 in loading RAD51 onto ssDNA during HR [Citation42,Citation43]. In line with these findings, Somyajit et al. showed that complexes of RAD51 paralogs bind to nascent DNA at stalled replication forks to prevent the formation of DSBs by protecting forks against MRE11 activity [Citation44]. The capacity to stabilize stalled replication forks appears very relevant in the context of PARP inhibition. Specifically, trapping of PARP enzymes onto DNA was shown to stall replication forks [Citation45], and PARP trapping lies at the basis of PARP inhibitor-induced cytotoxicity [Citation46]. Conversely, the degree to which cells can maintain replication fork stability was reported to determine PARP inhibitor sensitivity [Citation47].
2. HR-deficient cancers
Defects in DNA maintenance pathways, including DNA DSB repair, are a hallmark of cancer [Citation48]. Specifically, defective genome maintenance is coined an enabling feature of carcinogenesis, as it allows the accumulation of genetic errors. The subsequent genomic instability then facilitates the acquisition of tumor-promoting features. Defective DSB repair was also shown to result in mutations or chromosomal aberrations underlying carcinogenesis. Extensive research in the 1990s resulted in the identification of two breast cancer susceptibility genes, BRCA1 [Citation49,Citation50] and BRCA2 [Citation51,Citation52], of which heterozygous germline mutations result in an increased lifetime risk of developing breast and/or ovarian cancer. Besides the roles of BRCA1 and BRCA2 in HR described above, they exert additional functions related to DNA repair and cell cycle control to maintain genome stability [Citation21].
2.1. HR gene mutations
Germline BRCA1/2 mutations are predominantly linked to the development of breast and ovarian cancer, but they are also associated with an elevated risk for other cancer types, including pancreatic, prostate, and endometrial cancer [Citation53–Citation55]. Tumor onset in BRCA1/2 mutation carriers invariably involves loss of the remaining wild-type (wt) allele through somatic inactivation or loss-of-heterozygosity (LOH) and results in tumor cells that are HR defective [Citation56]. Besides mutation, epigenetic silencing of HR genes has also been shown to underpin defective HR in tumors. Specifically, the BRCA1 promoter is frequently hypermethylated in breast and ovarian cancer [Citation57,Citation58].
Importantly, not only germline BRCA1/2 mutations underlie HR deficiency in tumors, but somatic BRCA1/2 mutations have also been described [Citation59]. Also, mutations in other HR genes, such as PALB2 [Citation60,Citation61], RAD51 paralogs [Citation62], or ATM [Citation63], predispose to cancer development and may result in HR-defective tumors. In a cohort of patients with uterine serous carcinoma, different germline HR genes were found to be mutated [Citation64]. Furthermore, HR genes were shown to be mutated in lung, breast, intestinal, and skin cancer [Citation65]. Also, mutations in the cell cycle checkpoint gene CHEK2 were identified in breast cancer patients without a BRCA1/2 mutation (5.1%) when compared to healthy controls (1.1%) [Citation66,Citation67]. CHK2 is involved in BRCA1 phosphorylation upon DNA damage and has been implicated in controlling HR [Citation68,Citation69]. Whether the impact of the commonly occurring CHEK2 1100delC variant is strong enough to impact on HR repair and has therapeutic consequences needs to be established.
2.2. BRCA1/2 mutations in breast and ovarian cancer
Mutations in BRCA1 result in a ~65% lifetime risk for breast cancer development by the age of 70 years and a ~30–40% lifetime risk for ovarian cancer. For BRCA2 mutation carriers, the lifetime risk for breast cancer is around 50% at age 70 and ~10–15% for ovarian cancer [Citation70–Citation72]. The risk of cancer development depends on multiple factors, including the exact position of the mutation for both genes. Furthermore, somatic mutations in other genes such as TP53 [Citation73] or PTEN [Citation74] were suggested to influence BRCA1/2-related carcinogenesis.
Most breast cancers caused by BRCA1 mutations are ‘triple-negative’ breast cancers (TNBCs), which entails that they do not overexpress the estrogen receptor (ER), progesterone receptor, or the human epidermal growth factor receptor-2. TNBCs are characterized by aggressive growth and very limited targeted treatment options. In contrast, BRCA2-mutant breast cancers are mainly low-grade ER+ luminal tumors, which grow more slowly, and inhibition of signaling through the ER is one of the treatment options [Citation75,Citation76].
Ovarian tumors arising in BRCA1/2 mutation carriers are mainly high-grade serous carcinomas (HGSOCs) [Citation77]. Notably, when RNA expression profiles were examined, high levels of similarity were observed between BRCA1/2-related and non-BRCA1/2-related HGSOC, indicating that this subgroup is characterized by a high degree of genomic instability [Citation75]. Importantly, these observations suggest that inactivation of DNA repair is a common feature of serous ovarian cancer tumorigenesis. Indeed, genomic analysis by The Cancer Genome Atlas suggests that around half of HGSOCs are HR deficient, based on mutations in BRCA1/2 or mutations in other HR genes such as RAD51, ATM, CHEK2, PALB2, and MRE11 [Citation78].
2.3. Tumorigenesis and HR deficiency
BRCA1/2 genes have a tumor-suppressive function; heterozygous germline mutations in BRCA1 or BRCA2 predispose to cancer, in which cancer cells have lost the remaining wt allele and are fully HR defective. In apparent contradiction with this notion, HR deficiency caused by homozygous genetic inactivation of Brca1 [Citation79,Citation80], Brca2 [Citation81,Citation82], or Rad51 [Citation82,Citation83] causes early embryonic lethality in vivo, showing that HR is required for cell survival and development. The requirement for BRCA1 and BRCA2 extends beyond development since Brca1 or Brca2 knock-out mouse embryonic fibroblasts and blastocysts also display compromised viability in vitro [Citation80,Citation84]. Apparently, tumor cells that arise due to defective HR have developed mechanisms to cope with increased genomic instability. How these tumor cells survive and proliferate in the absence of HR is incompletely understood and was coined the ‘BRCA paradox’ [Citation85].
The enhanced rate of genomic aberrations induced by HR deficiency allows the accumulation of multiple secondary mutations, which support the survival of HR-deficient cells. Indeed, loss of HR leads to DNA damage accumulation and instigates a DDR, including transcriptional activation of p53 [Citation86], suggesting that the p53 signaling axis may preclude survival of HR-deficient cells. The observation that tumorigenesis in a Brca1 conditional mouse model was significantly accelerated by introducing a Tp53+/- mutation underscores the important role of p53 in BRCA1/2-associated tumors [Citation87]. Furthermore, a conditional mouse model with a CK14-driven Cre-mediated somatic loss of Brca1 and Tp53 resulted in a high incidence of mammary tumors that resemble human basal-like BRCA1 breast cancer [Citation88]. These data are in line with the human situation, in which TP53 is mutated in ~66% of BRCA1/2-related breast tumors [Citation89]. Combined, these observations explain the early embryonic death upon BRCA1/2 loss and show that HR-deficient cells cannot survive without a concomitant mutation in other genes, such as TP53. Interestingly, co-mutation of Tp53 only partially rescued the viability of cell cultures and mice lacking Brca1/2 [Citation90]. This suggests that other factors exist that promote BRCA1/2-related tumorigenesis and lead to survival of BRCA1/2-deficient tumor cells.
3. Therapeutic targeting of HR-deficient cancers
HR deficiency drives tumorigenesis but simultaneously provides an Achilles’ heel that can be exploited therapeutically. The absence of HR components is often correlated with improved therapeutic outcome [Citation1]. HR-deficient tumors are generally more sensitive to DNA damage that requires HR for repair, including platinum-induced DNA replication lesions.
3.1. Cross-linking agents and effectiveness
Different compounds can induce inter- or intra-strand cross-links (ICLs) which interfere with DNA replication. These drugs, including platinum-containing cytostatics, are widely used in various treatment settings for numerous cancer types including ovarian cancer. ICLs prevent separation of the DNA strands during replication and transcription and thus lead to stalled replication forks and stalled transcription [Citation91]. Besides template-based repair of DSBs, HR is also involved in the protection and restart of stalled replication forks and repair of ICLs (), right panel). This latter process is initiated by components of the Fanconi anemia (FA) pathway, which consists of multiple FA genes [Citation92]. Significant overlap exists between the components that function in HR and the FA pathways, including BRCA2 (FANCD1) [Citation93] and BRCA1 (FANC-S) [Citation94]. Germline mutations in FA genes lead to the FA syndrome, a very rare inherited disease. These patients are often diagnosed with cancer at an early age due to increased chromosomal instability [Citation95]. Of note, and in line with the repair function of FA genes, this syndrome is characterized by increased sensitivity to ICLs.
In epithelial ovarian carcinoma, both somatic and germline mutations in BRCA1 and BRCA2 are positively correlated with response to platinum-based treatment. A total of 14.9% of patients with a BRCA1/2 mutation had progressive disease within 6 months after primary treatment with platinum-based chemotherapy compared to 31.7% of patients with BRCA1/2 wt tumors [Citation96]. In addition, BRCA1/2 deficiency (either through mutation or loss of expression) is associated with improved progression-free survival (PFS) after platinum-based chemotherapy in serous ovarian cancer [Citation97]. Regardless of mutational status, decreased expression of BRCA1 was also positively correlated with response to cisplatin plus paclitaxel treatment [Citation98]. The increased response to chemotherapy in BRCA1/2-deficient ovarian cancers may underlie the fact that patients with germline BRCA1/2-mutated tumors have a better outcome in general (improved response rates and overall survival) [Citation99].
Whereas standard treatment of HGSOC is based on surgery and primary platinum-based chemotherapy, TNBCs in the past years were not consistently treated with platinum-based chemotherapy. A significant proportion of TNBCs are HR deficient, e.g. through BRCA1/2 mutations, and BRCA1/2-associated breast tumors have common characteristics with TNBCs in general [Citation100]. Rottenberg et al. have shown that spontaneous mammary mouse tumors induced by combined Brca1 and Tp53 inactivation resembled human BRCA1-associated breast cancer in humans [Citation101]. These Brca1−/−; Tp53−/− mouse tumors responded very well to cisplatin therapy and did not acquire resistance after five relapses, even though tumors were not completely eradicated [Citation101]. In a study with 190 TNBC patients, both the BRCA1/2 (16%) and the non-BRCA1/2-mutant tumors responded well to a neo-adjuvant combination therapy of carboplatin and docetaxel with pathologic complete responses in 59% and 56% of the cases, respectively [Citation102].
Low BRCA1 mRNA expression was found to be associated with increased cisplatin sensitivity in patients with TNBC [Citation103]. Finally, stage III breast cancer patients with a tumor of which the genomic pattern resembled BRCA1/2-mutated breast cancers and were thus classified as BRCA-like showed improved overall survival after high-dose platinum-containing chemotherapy (cyclophosphamide–thiotepa–carboplatin) compared to conventional 5-fluorouracil–epirubicin–cyclophosphamide (FE90C) therapy in a randomized controlled trial [Citation104]. These combined results have resulted in platinum-containing agents being increasingly included in standard chemotherapy regimens of TNBCs.
3.2. PARP inhibition
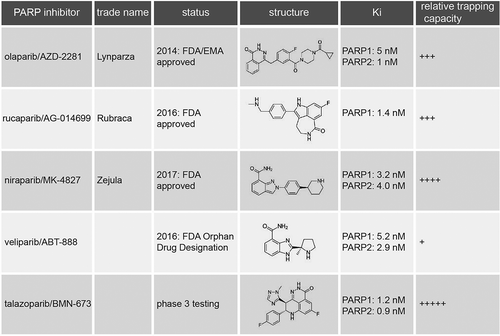
Based on the principle of synthetic lethality, new molecularly targeted therapeutic strategies have been developed for HR-deficient tumors, which interfere with remaining DNA repair pathways in the tumor [Citation105,Citation106]. PARP is an enzyme involved in base-excision repair (BER) which is used to repair SSBs [Citation107]. The first PARP inhibitor was developed in 1980 and was initially used to sensitize tumors to chemotherapy [Citation108]. In 2005, two seminal studies showed that BRCA1/2-mutated tumor cells were extremely sensitive to PARP inhibition, in contrast to BRCA1/2 heterozygote or wt cells due to synthetic lethality [Citation109,Citation110]. The developed small-molecule PARP inhibitors (KU0058684 and KU0058948) formed the basis for the first FDA-approved PARP inhibitor olaparib (AZD-2281, trade name: Lynparza, AstraZeneca Rubraca, Clovis Oncology Zejula, Tesaro) [Citation111,Citation112]. Very recently, two other PARP inhibitors were FDA approved, namely rucaparib (AG-014699, trade name: Rubraca) and niraparib (MK-4827, trade name: Zejula) [Citation113] ().
Figure 2. Overview of PARP inhibitors in clinical development. For each PARP inhibitor, various characteristics are indicated, including trade name, status in clinical development, chemical structure, dissociation constant (Ki) reflecting PARP1 catalytic inhibition, and capacity to trap PARP onto DNA.
3.2.1. Mechanisms of PARP inhibitor-induced cell death in HR-deficient tumor cells
Inhibition of the PARP enzyme results in insufficient repair and ensuring accumulation of SSBs, which are converted into DSBs during replication. Normal cells in BRCA1/2 mutation carriers still have a remaining BRCA1/2 allele and are therefore HR proficient. These cells can effectively repair DSBs and are only marginally affected by PARP inhibition. In contrast, tumor cells in which the remaining BRCA1/2 allele is lost are HR deficient and unable to effectively repair the DSBs induced by PARP inhibition and will ultimately undergo cell death. For this reason, the tumor specificity of PARP inhibitors is favorable when compared to traditional chemotherapeutic agents which target all dividing cells. Nevertheless, adverse side effects of PARP inhibition have been reported [Citation114,Citation115].
Recently, additional mechanisms of PARP inhibitor-induced cell death have been described. Besides interfering with SSB repair through inhibition of BER, PARP inhibitors can also trap the PARP enzyme onto the DNA to form protein:DNA complexes. These complexes behave like DNA inter-strand cross-links that interfere with DNA replication and require repair by the Fanconi pathway and HR machinery [Citation46,Citation116]. Again, for this mechanism to effectively induce cell death, lack of HR is required.
These studies also explain the observations that PARP inhibitors are most effective when PARP itself is abundantly present and that chemical PARP inhibition is more effective than removing PARP genetically [Citation45]. Many different PARP inhibitors have been described, all of which inactivate the PARP enzyme catalytically to a high degree (). However, these inhibitors differ in their capability to trap PARP onto DNA (). Notably, the cytotoxicity of the different PARP inhibitors is related to their trapping potential [Citation117]. Currently, the PARP inhibitor with highest trapping activity used in clinical studies is talazoparib (BMN-673), and this agent also has the highest single agent toxicity. The PARP trapping ability of talazoparib is a 100-fold higher than that of olaparib [Citation116,Citation118].
Of note, PARP1 was also shown to interact with NHEJ components. Specifically, PARP1 can bind to the NHEJ proteins Ku70/80 and DNA–PKcs and competes with Ku80 for repair of DSBs through an alternative NHEJ pathway [Citation119,Citation120]. In line with these observations, Patel et al. demonstrated that PARP inhibition leads to phosphorylation of DNA–PK substrates, thereby enhancing NHEJ activity in BRCA2-deficient cells [Citation121]. In the same study, inhibition of NHEJ through knockdown of Ku80 could increase the cell survival of BRCA2-deficient cells to PARP inhibition, suggesting that NHEJ repair of PARP inhibitor-induced DNA lesions contributes to the toxicity of PARP inhibitors. In line with this notion, inhibition of DNA–PK decreased the sensitivity of ATM- and BRCA1-deficient cancer cells to PARP inhibition [Citation121].
3.2.2. PARP inhibition in the clinic
In a phase I trial, only BRCA1/2 mutation carriers (n = 22) with different tumor types, including ovarian, breast, and prostate cancer, showed antitumor activity in response to olaparib monotherapy (63%) compared to non-mutation carriers [Citation112]. In the same study, adverse effects of olaparib monotherapy were observed that were mainly categorized as grade 1 or 2 and were, in general, less severe than those of classical chemotherapy. The observed presence of grade 3 adverse effects, such as myelosuppression and anemia, might be explained by long cancer history or pretreatment with chemotherapy regimens and be manageable by dose reduction or treatment interruption [Citation122].
A phase II trial included HGSOC patients who had received two or more platinum-based chemotherapy regimens and had a platinum-sensitive relapse [Citation123]. Patients were randomly assigned to olaparib monotherapy (n = 136) or placebo (n = 129), and PFS was significantly longer in the olaparib-treated group (median: 8.4 months) compared to patients treated with placebo (median: 4.8 months) [Citation123]. Most clinical trials with olaparib concern combination therapies with chemotherapeutic agents. For instance, in a randomized phase II trial, it was shown that olaparib combined with carboplatin and paclitaxel followed by olaparib monotherapy improves PFS in recurrent, platinum-sensitive HGSOC patients (median: 12.2 versus 9.6 months in chemotherapy alone), especially in patients with BRCA1/2 mutation (hazard ratio (HR): 0.21) [Citation124]. Maintenance monotherapy with olaparib significantly prolonged PFS versus placebo in patients with platinum-sensitive recurrent serous ovarian cancer, especially in patients with a BRCA1/2 mutation [Citation125]. Maintenance olaparib monotherapy in patients with BRCA1/2-mutated breast, ovarian, or fallopian tube tumors (n = 21) after combination chemotherapy with carboplatin and paclitaxel was well tolerated [Citation126] and has been approved by the European Medicines Agency (EMA) for this indication. In advanced, heavily pretreated, platinum-resistant ovarian cancer patients (n = 193), of whom 80% had germline BRCA1/2 mutations, olaparib monotherapy resulted in an objective response rate of 34% [Citation127], and this trial resulted in the FDA approval in this setting. Furthermore, in a multicenter phase II trial, heavily pretreated patients with a germline BRCA1/2 mutation (n = 298) were treated with olaparib monotherapy. This resulted in stable disease up to 8 weeks in 42% of the patients and an overall tumor response rate of 31.1% [Citation122]. Although olaparib showed responses as monotherapy, especially in BRCA1/2-mutant tumors, various studies have suggested that combination therapies are required to improve response rates [Citation128], likely at the cost of increased toxicity. In this context, numerous studies are ongoing.
3.2.3. Increasing the sensitivity for PARP inhibition
PARP inhibition is selectively cytotoxic in HR-deficient tumors. An approach to extend PARP eligibility to other HR-proficient tumors is to therapeutically induce temporary defects in HR. For instance, it was shown that HR is suppressed in multiple cancer cell lines under hypoxic conditions through downregulation of RAD51 [Citation129]. In addition, inhibition of vascular endothelial growth factor receptor 3 (VEGFR3) resulted in decreased expression of BRCA1 and BRCA2 in ovarian cancer cells [Citation130]. These data suggest that inhibiting angiogenesis can be used to enforce HR deficiency and improve responses to PARP inhibition. Indeed, when olaparib was combined with cediranib, a drug that targets the VEGFRs, improved PFS was observed in patients with platinum-sensitive recurrent ovarian, fallopian tube, or peritoneal tumors [Citation131]. Adverse effects of this combination therapy, however, were also increased when compared to olaparib treatment alone. Nevertheless, these adverse effects do not prevent current ongoing clinical trials.
Another family of enzymes involved in maintaining HR are phosphoinositide 3-kinases (PI3Ks), which are activated upon receptor signaling and have distinct functions in signal transduction pathways [Citation132]. The isoform PI3Kβ is found to be important for DSB sensing as it regulates recruitment of NBS1, a subunit of the MRN complex, to sites of DNA breaks [Citation133]. In line with these observations, Juvekar et al. showed that the PI3K and mitogen-activated protein kinase pathway were activated in a Brca1-mutated breast cancer mouse model, as judged by increased AKT and ERK phosphorylation [Citation134]. Conversely, PI3K class I inhibition using BKM-120 led to increased DNA damage and in combination with olaparib delayed in vivo tumor growth [Citation134]. Subsequently, PI3K inhibition in patients with BKM120 resulted in increased DNA damage in tumors, decreased levels of BRCA1 and BRCA2, and increased sensitivity of TNBCs to olaparib, even in tumors without a BRCA1/2 mutation [Citation135]. This combination may be valuable in other tumor types, as it also showed synergistic effects in human prostate cancer cell lines and in Pten/Tp53-mutaded mouse prostate tumors [Citation136]. Currently, an ongoing clinical trial combines BKM-120 with olaparib in TNBC and HGSOC patients (NCT01623349).
Another class of kinases that is essential for HR are CDKs. HR is strictly cell cycle regulated, which is governed by S and G2 CDKs, as explained above. In line with this notion, inhibition of CDK1 activity was shown to impair HR and to sensitize otherwise HR-proficient tumor cell lines for PARP inhibition [Citation137]. Furthermore, inhibition of multiple CDKs simultaneously using dinaciclib could overcome PARPi resistance in BRCA1/2-mutated TNBC cell lines and xenograft models by blocking the restored HR function [Citation138]. Dinaciclib is currently being assessed in combination with the PARP inhibitor veliparib in solid tumors (NCT01434316). Surprisingly, also a G1/S cyclin–CDK complex was found to be involved in HR regulation. Specifically, cyclin D, the non-catalytic partner of CDK4 and CDK6, appeared essential for HR, and this finding may open up additional possibilities to potentiate PARP inhibitor sensitivity [Citation139].
Finally, DNA repair through HR is inactivated in response to hyperthermia. The inhibition of HR shifts repair of DSBs to error-prone NHEJ and thereby sensitizes tumor cells to DNA damaging agents [Citation140]. Upon transient hyperthermia to 42.5°C, it was shown that BRCA2 is degraded in a proteasome-dependent fashion. Loss of BRCA2 lasts for several hours and functionally impairs HR. Consequently, tumor cells become sensitive to cisplatin, doxorubicin as well as PARP inhibitors in vitro and in vivo [Citation141]. This concept is currently being tested in a range of clinical trials, including a trial in head and neck cancer patients, testing the effects of hyperthermia on responses to the PARP inhibitor olaparib (Dutch Trial registry: NTR5842).
3.2.4. Resistance to PARP inhibitors
As with many molecularly targeted agents, resistance to PARP inhibitors is a clinical problem. Currently, different mechanisms underlying resistance to PARP inhibitor treatment have been described ().
Figure 3. PARP inhibitor resistance mechanisms. Various mechanisms for PARP inhibitor resistance are described. Secondary intragenic mutations (BRCA1/2), secondary mutations in other genes in BRCA1-mutant cancer cells or promotor translocations in BRCA1 may restore HR function. Secondary mutations in other genes may restore protection of stalled replication forks caused by BRCA2 inactivation.
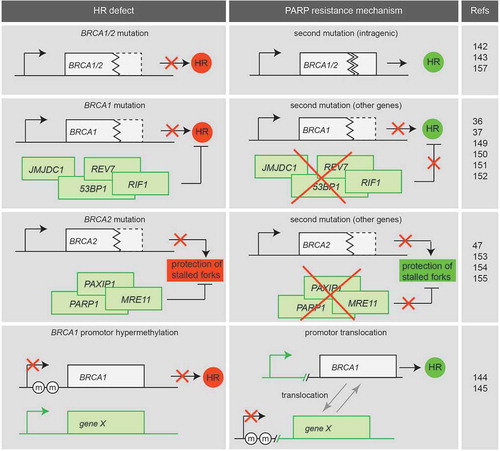
Secondary mutations or translocations may arise within the mutated BRCA1 or BRCA2 gene, restoring the reading frame of the affected gene. This was firstly described in breast and pancreatic cell lines in which secondary BRCA2 mutations restored the BRCA2 reading frame and resulted in cisplatin and PARP inhibitor resistance [Citation142]. The same research group reported that secondary mutations of BRCA1 also occur in platinum-resistant ovarian cancer with a BRCA1 mutation [Citation143]. In germline BRCA1/2-mutated ovarian cancer patients, secondary somatic mutations that restore BRCA1/2 were correlated with resistance to platinum-based chemotherapy [Citation144]. A mechanistically unrelated resistance mechanism was described for BRCA1-hypermethylated breast patient-derived xenograft (PDX) tumors, in which BRCA1 expression was restored through rearrangement of the BRCA1 locus, resulting in expression of BRCA1 from a different promoter [Citation145]. The loss of BRCA1 promoter methylation has already been described in chemotherapy-resistant ovarian cancer patients [Citation146].
The function of HR can also be restored by mutations in other genes. An important finding by Cao and coworkers described that loss of 53BP1 prevented the senescence and cell death induced by BRCA1 deficiency, both in vitro and in vivo [Citation147]. 53BP1 was originally identified as an activator of p53 in the DDRs [Citation148] and was later shown to promote NHEJ [Citation149]. Notably, 53BP1 inactivation partially restored HR in mouse embryonic stem cells with a conditional Brca1 knockout [Citation150]. Through this mechanism, loss of 53BP1 reversed the sensitivity of BRCA1-deficient cells to PARP inhibition [Citation150,Citation151]. Although these experiments were executed in mouse models, loss of 53BP1 may be a resistance mechanism to PARP inhibition in patients with BRCA1-mutant tumors [Citation152]. Indeed, altered expression of 53BP1 is commonly observed in BRCA1-mutated breast cancers.
Comparable observations were done for other NHEJ-promoting genes Rif1 and REV7 (also called Mad2L2). Mutation of these genes also rescued HR defects, promoted the cellular viability, and reversed PARP inhibitor sensitivity in BRCA1-deficient cells [Citation36,Citation153]. Additionally, it was shown that ubiquitylation and recruitment to DSBs of BRCA1, but not 53BP1, are regulated by the demethylase JMJD1C. Knockdown of JMJD1C resulted in increased RPA phosphorylation and accelerated formation of RAD51 foci upon irradiation. In BRCA1-depleted cells, knockdown of JMJD1C resulted in decreased sensitivity to PARP inhibition by olaparib and restored RAD51 foci formation [Citation37]. Furthermore, reduced expression of JMJD1C was found in a subset of invasive human breast cancers (26%), which suggests that JMJD1C is another player in PARP inhibitor resistance similar to 53BP1 and its cofactors.
Most of the above-described mechanisms reversed PARP inhibitor sensitivity and HR in BRCA1-mutant cancers, but not in BRCA2-mutant cancers. This probably reflects the upstream function of BRCA1 within the HR pathway, at the level of DNA-end resection initiation. BRCA2, by contrast, functions in RAD51 recruitment beyond the step of DNA-end resection. Recently, loss of PTIP (also known as PAX-interacting protein 1, encoded by the PAXIP1 gene) was described to rescue the lethality of Brca2-mutated embryonic mouse stem cells and caused PARP inhibitor resistance. However, PTIP inactivation did not restore HR, but rather lead to protection of replication forks through prevention of MRE11 recruitment to stalled replication forks [Citation47]. These data suggest that besides HR functionality, replication fork protection is critically involved in sensitivity to PARP inhibitors in BRCA2-deficient cancers.
As the cytotoxicity of PARP inhibition is dependent on the presence of its target PARP-1, it is suggested that decreased levels or activity of PARP-1 may interfere with PARP inhibitor response. The levels of PARP-1 were decreased in PARP inhibitor-resistant cell lines and increased activity of PARP-1 (as measured by PARylation) correlated to PARP inhibitor sensitivity [Citation154,Citation155]. Small nucleotide polymorphisms (SNPs) in the PARP1 gene may alter its function and activity and thereby influence the response to PARP inhibition [Citation156].
4. Patient selection for PARP inhibitor treatment
Currently, only serous ovarian cancer patients with proven germline or somatic BRCA1/2 mutations are eligible for treatment with olaparib or rucaparib (). In 2006, it was already suggested that PARP inhibition might be effective not only in tumors with BRCA1/2 mutations but also in tumors with loss of other HR components and in tumors beyond breast and ovarian cancer [Citation157]. Very recently, niraparib has also been approved by the FDA for treatment of recurrent fallopian tube or primary peritoneal cancer. Below, various techniques are described that can be used to facilitate patient selection for PARP inhibitor treatment.
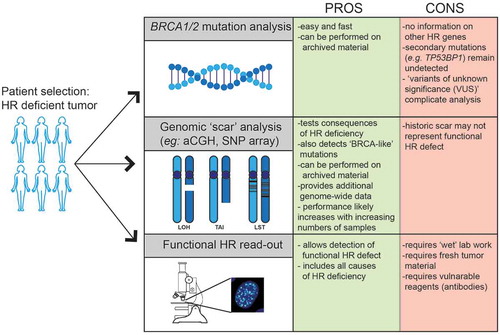
Figure 4. Patient selection for PARP inhibitor treatment. Currently, patients are selected for PARP inhibitor treatment based on BRCA1/2 mutation analysis. Additional techniques such as genomic scar analysis (e.g. array-CGH or DNA sequencing-based) or a functional HR read-out are being developed and could be included to better select patients with HR-deficient tumors. The advantages (PROS) and disadvantages (CONS) of each method are indicated.
4.1. Mutation analysis
BRCA1/2 mutational status and BRCA1 promoter methylation analysis of tumors will identify patients, likely to benefit from PARP inhibition. However, mutations in other HR genes might also result in HR deficiency and thus PARP inhibitor sensitivity, although these mutations are less frequently observed. Extending the panel of genes for mutational analysis might increase the selection of HR-deficient tumors, but for each of these genes, variants of unknown significance (VUS) occur which challenge clinical decision-making. In a study by Easton et al., 1433 VUS alleles in BRCA1 and BRCA2 were classified, of which the majority appeared to be of no significance in relation to cancer development [Citation144]. It was suggested that family history should play an important role in decision-making and prediction of cancer risk in patients with VUS alleles [Citation158]. Systemic approaches and combining big data sets is required to optimally classify the thousands of VUS alleles in BRCA1/2 and other HR genes to predict if these mutations predispose to cancer. In parallel, experimental models have been developed in which VUS alleles can be tested for functionality [Citation159]. Members of the global Evidence-based Network for the Interpretation of Germline Mutant Alleles (ENIGMA) consortium collaborate to better implement information on VUS alleles into clinical decision-making [Citation160].
Furthermore, secondary mutations either within the mutant BRCA1/2 alleles or in secondary genes that restore HR function have been described and may underlie resistance to platinum-based chemotherapy and PARP inhibitors [Citation161]. Profiling all these genes for mutations will make genetic screening increasingly complex.
4.2. Genomic scar analysis
Different approaches have been developed to discriminate between HR-proficient and HR-deficient tumors based on the landscape of the genomic tumor aberrations, referred to as a ‘genomic scar’ ().
To detect breast cancer tumors without BRCA1/2 mutations, but with a similar phenotype, a classifier was developed based on tumor profiles with array-comparative genomic hybridization (CGH) using a set of BRCA1-mutated breast tumors as well as control breast tumors [Citation162]. In a group of 48 patients from families with hereditary breast and ovarian cancer, two tumors with a ‘BRCA1-like’ array-CGH profile but without BRCA1/2 germline mutation were detected. Furthermore, this classifier predicted response to genotoxic agents with improved outcome of ‘BRCA1-like’ tumors (based on the array-CGH profile) to platinum-based chemotherapy in stage III breast cancer patients [Citation163]. Of note, this technique might not only be useful for prediction but also to give insight into the significance of certain VUS alleles and identify compensatory genomic alterations that facilitate cellular survival in the absence of HR.
In a recent study by Davies et al., whole-genome profiling was applied to 24 breast tumors with a germline BRCA1/2 mutation and results were compared to sporadic breast cancer samples to develop an algorithm that can differentiate between these groups [Citation164]. Included parameters were based on indels, base-substitutions, and rearrangements. In different additional cohorts of breast, pancreatic, and ovarian cancer, tumors with a BRCA1/2 deficiency were identified when the developed algorithm (named ‘HRDetect’) was applied. These tumors harbored either biallelic germline or somatic mutations in BRCA1 or BRCA2 or promoter hypermethylation of BRCA1 combined with a loss of the second allele. Importantly, also tumors without genetic alterations in BRCA1/2 were identified, illustrating that sequence analysis for BRCA1/2 alone is insufficient to detect all tumors with an HR-deficient phenotype [Citation164].
Myriad Genetics has developed a homologous recombination deficiency (HRD) test to identify patients that could benefit from PARP inhibitor treatment (termed ‘MyChoice’ test). This test includes a genetic and phenotypic analysis of formalin-fixed, paraffin-embedded tumor tissue collected by biopsy or surgery. Genes associated with HR deficiency are sequenced, including BRCA1/2 as well as others [Citation165]. As this analysis cannot identify tumors with epigenetically silenced HR genes and other yet unknown causes of HR deficiency, tumor tissue is also analyzed at a phenotypic level for three features of genomic instability. These characteristics include large-scale transitions, clustering of LOH, and assessing the telomeric allelic imbalance rates (reviewed in [Citation166]). The tumors are assigned a combined HRD score based on these three characteristics. It has been shown that this phenotypic HRD score strongly correlates with a BRCA1/2 deficiency in different types of breast tumors [Citation167]. The combination of mutational analysis and HRD score gives a better prediction of HR compared to mutational status alone. Currently, based on clinical trials, the MyChoice test identifies twice as many patients that may benefit from PARP inhibitor treatment and platinum-based chemotherapy compared to selection by BRCA1/2 mutational analyses alone for both breast and ovarian cancer (NCT01847274) [Citation168]. Included patients without BRCA1/2 mutations but with high HRD score appear to show a favorable response to platinum-based therapy in TNBC [Citation169]. However, not all HR-deficient tumors based on mutational analysis or genomic scarring will be sensitive to PARP inhibition. These tumors are highly genomically unstable and may, therefore, develop secondary mutations that restore HR function or result in PARP inhibitor resistance as described above.
Genomic scar analysis is performed on tumor specimens taken prior to treatment. The disadvantage of this approach is that it provides a historic representation of the genetic aberrations in the tumor, but does not reflect current HR deficiency as for instance influenced by secondary mutations. Additional biomarkers or functional assays to determine whether HR is still defective or possibly restored will, therefore, provide better insight.
4.3. Functional HR read-out – RAD51 foci formation
The essential last step in HR repair is RAD51 loading, and its functionality can be visualized by foci formation analysis [Citation170]. Mechanistically, BRCA2 is required for RAD51 foci formation upon DSBs induced by ionizing radiation (IR) [Citation171]. As RAD51 is the effector in HR, lack of BRCA2, but also upstream HR defects in components such as BRCA1 or PALB2, results in the absence of RAD51 foci formation. The formation of RAD51 foci is, therefore, a functional read-out for HR deficiency ().
To determine the ability of cells to repair DSBs by HR, different in vitro or ex vivo models have been used to assess the formation of irradiation-induced RAD51 foci. In ovarian cancer cell lines and in PDX models from omental tumors, ex vivo assessed irradiation-induced RAD51 foci correlated with response to the PARP inhibitor veliparib (ABT-888) [Citation172]. In primary cultures of ascites from patients with epithelial ovarian cancer, a correlation was found between the response to PARP inhibition (AG14699) and decreased RAD51 foci formation, although in this study RAD51 foci formation was determined at 24 h after treatment with AG14699, rather than at short-term interval upon IR [Citation173].
It is important to consider that different sources of DSBs, such as IR versus chemical compounds, may lead to a different time frame in which RAD51 foci appear. It has been shown that efficient DNA repair, and thus formation of RAD51 foci, in response to irradiation is optimal after 2 h [Citation174]. Counting RAD51 foci at 24 h after treatment may, therefore, lead to an overestimation of tumors that are HR deficient. Furthermore, both studies did not discriminate between cells in different phases of the cell cycle [Citation172,Citation173]. Since HR only occurs in S and G2 of the cell cycle, RAD51 foci will only appear in a subset of tumor cells. If RAD51 foci are counted in cells regardless of the cell cycle phase, it may result in false-negative results, for instance in tumor samples that contain a high percentage of non-proliferating cells. The appearance of false negatives was indeed the case in Mukhopadhyay et al. To reliably determine HR functionality, a cell cycle or proliferation markers should be included. Geminin, for instance, is a nuclear protein that is present during S and G2 phase to coordinate replication and can, therefore, be used as cell cycle marker [Citation175]. Taking geminin into account as a cell cycle marker provides an additional check to determine whether the ex vivo cultures are still proliferating. Naipal et al. determined the presence of RAD51 foci upon irradiation in geminin-positive cells of ex vivo breast cancer tissue samples. In this study, 11% of samples were HR deficient, and defective RAD51 foci formation correlated with TNBC status [Citation176].
In another study with fresh tumor samples of breast cancer patients, ex vivo RAD51 foci formation was assessed, and 22% of tumors were found to be RAD51 deficient and thus HR defective. Subsequently, biallelic inactivation of different HR genes was detected by sequencing and could explain almost 90% of the RAD51-foci devoid of tumors [Citation177].
Graeser et al. assessed RAD51 foci formation in biopsies of patients taken at 24 h after neo-adjuvant chemotherapy. RAD51 foci were assessed in geminin-positive cells, and HR deficiency was found in 26% of the tumors, which were again enriched for TNBC status [Citation178]. Also, low levels of RAD51 foci correlated with pathologic complete response to anthracycline-based chemotherapy (33%), when compared to tumors that were HR proficient (3%). Different approaches to counting RAD51 foci in multiple studies, such as the time point after irradiation, may explain the variety in percentages of HR-deficient tumors.
5. Conclusion
Repair of DNA DSBs and collapsed replication forks depends on HR for efficient resolution. Defective HR, such as caused by cancer-associated mutations in BRCA1, BRCA2, or related HR genes, leads to genomic instability and facilitates tumor progression. Yet, HR defects come with acquired sensitivity to DNA damaging agents, including PARP inhibitors. Current patient inclusion is largely based on BRCA1/2 mutational analysis. However, BRCA1/2 mutational analysis is likely not sufficient to include all HR-deficient tumors, and conversely, some BRCA1/2 mutant cancers may be HR proficient, due to secondary mutations. The restoration of HR underlies one of the mechanisms by which tumors become resistant to PARP inhibition, especially in BRCA1-mutant tumors. Restoration of replication fork stability appears to be another mechanism of PARP inhibitor resistance, especially in BRCA2-deficient tumors. Development of functional HR deficiency tests may more reliably identify patients who may benefit from PARP inhibition. Functional assays in preclinical testing have correlated RAD51 foci formation with clinical parameters and response to DNA damaging agents [Citation178]. A clinical trial designed to determine whether ex vivo RAD51 foci formation can predict responses to PARP inhibition in multiple tumor types (NCT03044795) is due to commence soon. Additionally, functional testing at the time of resistance to PARP inhibitor therapy may aid in yielding a better understanding of the mechanisms of acquired PARP resistance.
6. Expert opinion
PARP inhibition in HR-deficient cancers is the prototypical example of personalized medicine, based on synthetic lethality. Currently, three PARP inhibitors have been approved by the FDA, and olaparib has been approved by the EMA for BRCA1/2-mutated ovarian cancer. Several other PARP inhibitors are in clinical development. Increasingly, it appears that the ability to trap the PARP enzyme onto DNA is important for cytotoxic effects, in addition to their ability to catalytically inhibit PARP.
To optimally implement PARP inhibitors in cancer treatment, selection of the patients with most suitable tumors is key. Genetic testing for BRCA1/2 mutation remains a powerful approach, but might miss a significant number of HR-deficient tumors, harboring BRCA1 promoter hypermethylation or mutations in other HR genes. Identification of such tumors is challenged by the multitude of genes involved in HR and by our limited understanding of the contribution of each of these genes.
Ideally, selection of eligible patients for PARP inhibitor treatment involves a test that measures downstream consequences of defective HR. Existing tests are based on genomic platforms such as array-CGH, SNP arrays, or deep sequencing-based analysis and display genomic ‘scars’ induced by HR deficiency. Using algorithms, genomic scars can be identified that resemble those of BRCA1/2-mutant cancers and predict HR deficiency, regardless of the underlying gene mutation. These assays will grow increasingly reliable, with growing numbers of samples analyzed.
As with other targeted anticancer agents, acquired resistance to PARP inhibitors occurs. Increasingly, the genetic events that may underlie resistance are uncovered and could be included in decision-making for PARP inhibitor treatment. Over the last years, multiple genetic alterations have been described that can rescue defective HR and thereby render tumor cells insensitive to PARP inhibitors. Importantly, genomic scars represent historic events and may not reflect current HR deficiency when such secondary mutations have occurred.
To address this issue, assays are required that functionally interrogate HR functionality. In this context, fresh tumor samples can be prepared and analyzed for their ability to induce focus formation of the HR component RAD51 or related downstream HR components. Although these assays are technically feasible and require fresh tumor material, they theoretically would be able to include all HR-deficient tumors, beyond breast and ovarian cancer. Most of the tumor tissue in studies that assess RAD51 foci formation is irradiated as a model to induce DSBs. In an ideal situation, PARP inhibitors are employed instead of irradiation, as they instigate the most relevant type of DNA lesions and activate the relevant DNA repair pathway. Furthermore, some tumors contain a small portion of actively proliferating cells. Since RAD51 foci formation can only be functional in proliferating cells, it may turn out to be challenging to assess sufficient amounts of cycling tumor cells. Finally, different approaches may need to be tested to keep tumor tissues viable for the duration of the ex vivo procedure.
Recent insight has also shown that PARP inhibitor sensitivity is associated with the ability of tumors to stabilize stalled replication forks, a mechanism that also involves HR components. The ideal functional assay to test PARP inhibitor eligibility therefore not only includes RAD51 foci formation but also involves the ability of cancer cells to maintain replication fork stability. Various technical hurdles will need to be overcome to implement such functional assays clinically.
Combined, PARP inhibitors may provide clinical benefit for various cancers, beyond BRCA1/2-mutant ovarian cancers. To facilitate patient selection for PARP inhibitors, additional tests beyond BRCA1/2 mutational analysis should be employed, ranging from genetic analysis to functional assays in fresh tumor tissue. In the coming years, accurate ways to select patients for PARP inhibitor treatment will be assessed in the context of clinical trials. As more PARP inhibitor resistance mechanisms are being discovered, it is important to be able to detect if resistance mechanisms are active in the tumor to efficiently adapt the treatment with other treatment regimens, such as immunotherapies.
Article highlights
DNA double strand breaks and collapsed replication forks can be repaired by homologous recombination (HR)
Mutations in HR genes predispose subjects to the development of cancer
Patients with HR-deficient tumors benefit from platinum-based chemotherapy and PARP inhibitor treatment
Restoration of HR function and protection of replication forks underlies therapy resistance
The number of patients that benefit from PARP inhibitors may increase if patient selection is based on ‘genomic scar’ analysis or functional HR deficiency tests in tumor tissue, instead of BRCA1/2 mutation analysis
This box summarizes key points contained in the article.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Acknowledgments
We thank Dik van Gent, Titia Meijer, Agnes Jager, Maaike Vreeswijk, Jos Jonkers for fruitful discussions.
Additional information
Funding
References
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009 Oct 22;461(7267):1071–1078.
- Matsuoka S, Ballif BA, Smogorzewska A, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007 May 25;316(5828):1160–1166.
- Bassing CH, Swat W, Alt FW. The mechanism and regulation of chromosomal V(D)J recombination. Cell. 2002 Apr;109(Suppl):S45–55.
- Fugmann SD, Lee AI, Shockett PE, et al. The RAG proteins and V(D)J recombination: complexes, ends, and transposition. Annu Rev Immunol. 2000;18:495–527.
- Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014 Jan;16(1):2–9.
- Aymard F, Bugler B, Schmidt CK, et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat Struct Mol Biol. 2014 Apr;21(4):366–374.
- Lieber MR. The mechanism of human nonhomologous DNA end joining. J Biol Chem. 2008 Jan 4;283(1):1–5.
- Pitcher RS, Wilson TE, Doherty AJ. New insights into NHEJ repair processes in prokaryotes. Cell Cycle. 2005 May;4(5):675–678.
- Ma Y, Lu H, Schwarz K, et al. Repair of double-strand DNA breaks by the human nonhomologous DNA end joining pathway: the iterative processing model. Cell Cycle. 2005 Sep;4(9):1193–1200.
- Radhakrishnan SK, Jette N, Lees-Miller SP. Non-homologous end joining: emerging themes and unanswered questions. DNA Repair (Amst). 2014 May;17:2–8.
- Bennardo N, Cheng A, Huang N, et al. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. Plos Genet. 2008 Jun 27;4(6):e1000110.
- Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol. 2010 Mar;11(3):196–207.
- Wyman C, Kanaar R. DNA double-strand break repair: all’s well that ends well. Annu Rev Genet. 2006;40:363–383.
- Johnson RD, Jasin M. Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. Embo J. 2000 Jul 3;19(13):3398–3407.
- Wu X, Ranganathan V, Weisman DS, et al. ATM phosphorylation of Nijmegen breakage syndrome protein is required in a DNA damage response. Nature. 2000 May 25;405(6785):477–482.
- Zhao S, Weng YC, Yuan SS, et al. Functional link between ataxia-telangiectasia and Nijmegen breakage syndrome gene products. Nature. 2000 May 25;405(6785):473–477.
- Anand R, Ranjha L, Cannavo E, et al. Phosphorylated CtIP functions as a co-factor of the MRE11-RAD50-NBS1 endonuclease in DNA end resection. Mol Cell. 2016 Dec 1;64(5):940–950.
- Garcia V, Phelps SE, Gray S, et al. Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature. 2011 Oct 16;479(7372):241–244.
- Nimonkar AV, Genschel J, Kinoshita E, et al. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011 Feb 15;25(4):350–362.
- Sturzenegger A, Burdova K, Kanagaraj R, et al. DNA2 cooperates with the WRN and BLM RecQ helicases to mediate long-range DNA end resection in human cells. J Biol Chem. 2014 Sep 26;289(39):27314–27326.
- Gudmundsdottir K, Ashworth A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene. 2006 Sep 25;25(43):5864–5874.
- Suwaki N, Klare K, Tarsounas M. RAD51 paralogs: roles in DNA damage signalling, recombinational repair and tumorigenesis. Semin Cell Dev Biol. 2011 Oct;22(8):898–905.
- Lok BH, Carley AC, Tchang B, et al. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene. 2013 Jul 25;32(30):3552–3558.
- Badie S, Liao C, Thanasoula M, et al. RAD51C facilitates checkpoint signaling by promoting CHK2 phosphorylation. J Cell Biol. 2009 May 18;185(4):587–600.
- Swagemakers SM, Essers J, De Wit J, et al. The human RAD54 recombinational DNA repair protein is a double-stranded DNA-dependent ATPase. J Biol Chem. 1998 Oct 23;273(43):28292–28297.
- Bugreev DV, Mazina OM, Mazin AV. Rad54 protein promotes branch migration of Holliday junctions. Nature. 2006 Aug 3;442(7102):590–593.
- Moudry P, Watanabe K, Wolanin KM, et al. TOPBP1 regulates RAD51 phosphorylation and chromatin loading and determines PARP inhibitor sensitivity. J Cell Biol. 2016 Feb 1;212(3):281–288.
- Pace P, Mosedale G, Hodskinson MR, et al. Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science. 2010 Jul 9; 329(5988):219–223.
- Ceccaldi R, Liu JC, Amunugama R, et al. Homologous-recombination-deficient tumours are dependent on poltheta-mediated repair. Nature. 2015 Feb 12;518(7538):258–262.
- Kent T, Chandramouly G, McDevitt SM, et al. Mechanism of microhomology-mediated end-joining promoted by human DNA polymerase theta. Nat Struct Mol Biol. 2015 Mar;22(3):230–237.
- Sartori AA, Lukas C, Coates J, et al. Human CtIP promotes DNA end resection. Nature. 2007 Nov 22;450(7169):509–514.
- Yu X, Chen J. DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol Cell Biol. 2004 Nov;24(21):9478–9486.
- Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell. 2013 Mar 7;49(5):872–883.
- Isono M, Niimi A, Oike T, et al. BRCA1 directs the repair pathway to homologous recombination by promoting 53BP1 dephosphorylation. Cell Rep. 2017 Jan 10;18(2):520–532.
- Boersma V, Moatti N, Segura-Bayona S, et al. MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5ʹ end resection. Nature. 2015 May 28;521(7553):537–540.
- Xu G, Chapman JR, Brandsma I, et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature. 2015 May 28;521(7553):541–544.
- Watanabe S, Watanabe K, Akimov V, et al. JMJD1C demethylates MDC1 to regulate the RNF8 and BRCA1-mediated chromatin response to DNA breaks. Nat Struct Mol Biol. 2013 Dec;20(12):1425–1433.
- Tkac J, Xu G, Adhikary H, et al. HELB is a feedback inhibitor of DNA end resection. Mol Cell. 2016 Feb 4;61(3):405–418.
- Chroma K, Mistrik M, Moudry P, et al. Tumors overexpressing RNF168 show altered DNA repair and responses to genotoxic treatments, genomic instability and resistance to proteotoxic stress. Oncogene. 2016 Nov 14.
- Densham RM, Garvin AJ, Stone HR, et al. Human BRCA1-BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat Struct Mol Biol. 2016 Jul;23(7):647–655.
- Yata K, Lloyd J, Maslen S, et al. Plk1 and CK2 act in concert to regulate Rad51 during DNA double strand break repair. Mol Cell. 2012 Feb 10;45(3):371–383.
- Schlacher K, Christ N, Siaud N, et al. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011 May 13; 145(4):529–542.
- Schlacher K, Wu H, Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell. 2012 Jul 10;22(1):106–116.
- Somyajit K, Saxena S, Babu S, et al. Mammalian RAD51 paralogs protect nascent DNA at stalled forks and mediate replication restart. Nucleic Acids Res. 2015 Nov 16;43(20):9835–9855.
- Strom CE, Johansson F, Uhlen M, et al. Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res. 2011 Apr;39(8):3166–3175.
- Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012 Nov 1;72(21):5588–5599.
- Ray Chaudhuri A, Callen E, Ding X, et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature. 2016 Jul 20;535(7612):382–387.
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011 Mar 4;144(5):646–674.
- Futreal PA, Liu Q, Shattuck-Eidens D, et al. BRCA1 mutations in primary breast and ovarian carcinomas. Science. 1994 Oct 7;266(5182):120–122.
- Miki Y, Swensen J, Shattuck-Eidens D, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994 Oct 7;266(5182):66–71.
- Tavtigian SV, Simard J, Rommens J, et al. The complete BRCA2 gene and mutations in chromosome 13q-linked kindreds. Nat Genet. 1996 Mar;12(3):333–337.
- Wooster R, Bignell G, Lancaster J, et al. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995 Dec 21-28;378(6559):789–792.
- Iqbal J, Ragone A, Lubinski J, et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br J Cancer. 2012 Dec 4;107(12):2005–2009.
- Thompson D, Easton DF. Breast cancer linkage consortium. Cancer incidence in BRCA1 mutation carriers. J Natl Cancer Inst. 2002 Sep 18;94(18):1358–1365.
- Mersch J, Jackson M, Park M, et al. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer. 2015 Jul 15;121(14):2474–2475.
- Osorio A, De La Hoya M, Rodriguez-Lopez R, et al. Loss of heterozygosity analysis at the BRCA loci in tumor samples from patients with familial breast cancer. Int J Cancer. 2002 May 10;99(2):305–309.
- Esteller M, Silva JM, Dominguez G, et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst. 2000 Apr 5;92(7):564–569.
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011 Jun 29;474(7353):609–615.
- Moschetta M, George A, Kaye SB, et al. BRCA somatic mutations and epigenetic BRCA modifications in serous ovarian cancer. Ann Oncol. 2016 Aug;27(8):1449–1455.
- Ramus SJ, Song H, Dicks E, et al. Germline mutations in the BRIP1, BARD1, PALB2, and NBN genes in women with ovarian cancer. J Natl Cancer Inst. 2015 Aug 27;107(11). Print 2015 Nov. doi:10.1093/jnci/djv214.
- Antoniou AC, Casadei S, Heikkinen T, et al. Breast-cancer risk in families with mutations in PALB2. N Engl J Med. 2014 Aug 7;371(6):497–506.
- Song H, Dicks E, Ramus SJ, et al. Contribution of germline mutations in the RAD51B, RAD51C, and RAD51D genes to ovarian cancer in the population. J Clin Oncol. 2015 Sep 10;33(26):2901–2907.
- Minion LE, Dolinsky JS, Chase DM, et al. Hereditary predisposition to ovarian cancer, looking beyond BRCA1/BRCA2. Gynecol Oncol. 2015 Apr;137(1):86–92.
- Frimer M, Levano KS, Rodriguez-Gabin A, et al. Germline mutations of the DNA repair pathways in uterine serous carcinoma. Gynecol Oncol. 2016 Apr;141(1):101–107.
- Chae YK, Anker JF, Carneiro BA, et al. Genomic landscape of DNA repair genes in cancer. Oncotarget. 2016 Apr 26;7(17):23312–23321.
- Meijers-Heijboer H, Van Den Ouweland A, Klijn J, et al. Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat Genet. 2002 May;31(1):55–59.
- Vahteristo P, Bartkova J, Eerola H, et al. A CHEK2 genetic variant contributing to a substantial fraction of familial breast cancer. Am J Hum Genet. 2002 Aug;71(2):432–438.
- Lee JS, Collins KM, Brown AL, et al. hCds1-mediated phosphorylation of BRCA1 regulates the DNA damage response. Nature. 2000 Mar 9;404(6774):201–204.
- Zhang J, Willers H, Feng Z, et al. Chk2 phosphorylation of BRCA1 regulates DNA double-strand break repair. Mol Cell Biol. 2004 Jan;24(2):708–718.
- Antoniou A, Pharoah PD, Narod S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003 May;72(5):1117–1130.
- Easton DF, Ford D, Bishop DT. Breast and ovarian cancer incidence in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Am J Hum Genet. 1995 Jan;56(1):265–271.
- Mavaddat N, Peock S, Frost D, et al. Cancer risks for BRCA1 and BRCA2 mutation carriers: results from prospective analysis of EMBRACE. J Natl Cancer Inst. 2013 Jun 5;105(11):812–822.
- Shive HR, West RR, Embree LJ, et al. BRCA2 and TP53 collaborate in tumorigenesis in zebrafish. Plos One. 2014 Jan 29;9(1):e87177.
- van der Groep P, van der Wall E, van Diest PJ. Pathology of hereditary breast cancer. Cell Oncol (Dordr). 2011 Apr;34(2):71–88.
- Jazaeri AA, Yee CJ, Sotiriou C, et al. Gene expression profiles of BRCA1-linked, BRCA2-linked, and sporadic ovarian cancers. J Natl Cancer Inst. 2002 Jul 3;94(13):990–1000.
- Lakhani SR, van de Vijver MJ, Jacquemier J, et al. The pathology of familial breast cancer: predictive value of immunohistochemical markers estrogen receptor, progesterone receptor, HER-2, and p53 in patients with mutations in BRCA1 and BRCA2. J Clin Oncol. 2002 May 1;20(9):2310–2318.
- Moslehi R, Chu W, Karlan B, et al. BRCA1 and BRCA2 mutation analysis of 208 Ashkenazi Jewish women with ovarian cancer. Am J Hum Genet. 2000 Apr;66(4):1259–1272.
- Helleday T, Petermann E, Lundin C, et al. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008 Mar;8(3):193–204.
- Gowen LC, Johnson BL, Latour AM, et al. Brca1 deficiency results in early embryonic lethality characterized by neuroepithelial abnormalities. Nat Genet. 1996 Feb;12(2):191–194.
- Hakem R, De La Pompa JL, Sirard C, et al. The tumor suppressor gene Brca1 is required for embryonic cellular proliferation in the mouse. Cell. 1996 Jun 28;85(7):1009–1023.
- Suzuki A, De La Pompa JL, Hakem R, et al. Brca2 is required for embryonic cellular proliferation in the mouse. Genes Dev. 1997 May 15;11(10):1242–1252.
- Sharan SK, Morimatsu M, Albrecht U, et al. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature. 1997 Apr 24;386(6627):804–810.
- Ludwig T, Chapman DL, Papaioannou VE, et al. Targeted mutations of breast cancer susceptibility gene homologs in mice: lethal phenotypes of Brca1, Brca2, Brca1/Brca2, Brca1/p53, and Brca2/p53 nullizygous embryos. Genes Dev. 1997 May 15;11(10):1226–1241.
- Patel KJ, Yu VP, Lee H, et al. Involvement of Brca2 in DNA repair. Mol Cell. 1998 Feb;1(3):347–357.
- Elledge SJ, Amon A. The BRCA1 suppressor hypothesis: an explanation for the tissue-specific tumor development in BRCA1 patients. Cancer Cell. 2002 Mar;1(2):129–132.
- Hirao A, Kong YY, Matsuoka S, et al. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science. 2000 Mar 10;287(5459):1824–1827.
- Brodie SG, Xu X, Qiao W, et al. Multiple genetic changes are associated with mammary tumorigenesis in Brca1 conditional knockout mice. Oncogene. 2001 Nov 8;20(51):7514–7523.
- Liu X, Holstege H, van der Gulden H, et al. Somatic loss of BRCA1 and p53 in mice induces mammary tumors with features of human BRCA1-mutated basal-like breast cancer. Proc Natl Acad Sci U S A. 2007 Jul 17;104(29):12111–12116.
- Crook T, Brooks LA, Crossland S, et al. p53 mutation with frequent novel condons but not a mutator phenotype in BRCA1- and BRCA2-associated breast tumours. Oncogene. 1998 Oct 1;17(13):1681–1689.
- Evers B, Jonkers J. Mouse models of BRCA1 and BRCA2 deficiency: past lessons, current understanding and future prospects. Oncogene. 2006 Sep 25;25(43):5885–5897.
- Deans AJ, West SC. DNA interstrand crosslink repair and cancer. Nat Rev Cancer. 2011 Jun 24;11(7):467–480.
- Kee Y, D’Andrea AD. Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev. 2010 Aug 15;24(16):1680–1694.
- Howlett NG, Taniguchi T, Olson S, et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002 Jul 26;297(5581):606–609.
- Sawyer SL, Tian L, Kahkonen M, et al.; University of Washington Centre for Mendelian Genomics. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov. 2015 Feb;5(2):135–142.
- Niedernhofer LJ, Lalai AS, Hoeijmakers JH. Fanconi anemia (cross)linked to DNA repair. Cell. 2005 Dec 29;123(7):1191–1198.
- Alsop K, Fereday S, Meldrum C, et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group. J Clin Oncol. 2012 Jul 20;30(21):2654–2663.
- Hennessy BT, Timms KM, Carey MS, et al. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. J Clin Oncol. 2010 Aug 1;28(22):3570–3576.
- Lesnock JL, Darcy KM, Tian C, et al. BRCA1 expression and improved survival in ovarian cancer patients treated with intraperitoneal cisplatin and paclitaxel: a gynecologic oncology group study. Br J Cancer. 2013 Apr 2;108(6):1231–1237.
- Tan DS, Rothermundt C, Thomas K, et al. “BRCAness” syndrome in ovarian cancer: a case-control study describing the clinical features and outcome of patients with epithelial ovarian cancer associated with BRCA1 and BRCA2 mutations. J Clin Oncol. 2008 Dec 1;26(34):5530–5536.
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012 Oct 4;490(7418):61–70.
- Rottenberg S, Nygren AO, Pajic M, et al. Selective induction of chemotherapy resistance of mammary tumors in a conditional mouse model for hereditary breast cancer. Proc Natl Acad Sci U S A. 2007 Jul 17;104(29):12117–12122.
- Sharma P, Lopez-Tarruella S, Garcia-Saenz JA, et al. Efficacy of neoadjuvant carboplatin plus docetaxel in triple-negative breast cancer: combined analysis of two cohorts. Clin Cancer Res. 2017 Feb 1;23(3):649–657.
- Silver DP, Richardson AL, Eklund AC, et al. Efficacy of neoadjuvant cisplatin in triple-negative breast cancer. J Clin Oncol. 2010 Mar 1;28(7):1145–1153.
- Vollebergh MA, Lips EH, Nederlof PM, et al. Genomic patterns resembling BRCA1- and BRCA2-mutated breast cancers predict benefit of intensified carboplatin-based chemotherapy. Breast Cancer Res. 2014 May 15;16(3):R47.
- Lucchesi JC. Synthetic lethality and semi-lethality among functionally related mutants of Drosophila melanogaster. Genetics. 1968 May;59(1):37–44.
- Hartwell LH, Szankasi P, Roberts CJ, et al. Integrating genetic approaches into the discovery of anticancer drugs. Science. 1997 Nov 7;278(5340):1064–1068.
- Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008 Jan;18(1):27–47.
- Purnell MR, Whish WJ. Novel inhibitors of poly(ADP-ribose) synthetase. Biochem J. 1980 Mar 1;185(3):775–777.
- Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005 Apr 14;434(7035):917–921.
- Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005 Apr 14;434(7035):913–917.
- Gelmon KA, Tischkowitz M, Mackay H, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011 Sep;12(9):852–861.
- Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009 Jul 9;361(2):123–134.
- Mirza MR, Monk BJ, Herrstedt J, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016 Dec 1;375(22):2154–2164.
- Lord CJ, Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Science. 2017 Mar 17;355(6330):1152–1158.
- Brown JS, Kaye SB, Yap TA. PARP inhibitors: the race is on. Br J Cancer. 2016 Mar 29;114(7):713–715.
- Murai J, Huang SY, Renaud A, et al. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther. 2014 Feb;13(2):433–443.
- Pommier Y, O’Connor MJ, De Bono J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med. 2016 Oct 26;8(362):362ps17.
- Shen Y, Rehman FL, Feng Y, et al. BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin Cancer Res. 2013 Sep 15;19(18):5003–5015.
- Wang M, Wu W, Wu W, et al. PARP-1 and ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34(21):6170–6182.
- Li B, Navarro S, Kasahara N, et al. Identification and biochemical characterization of a Werner’s syndrome protein complex with Ku70/80 and poly(ADP-ribose) polymerase-1. J Biol Chem. 2004 Apr 2;279(14):13659–13667.
- Patel AG, Sarkaria JN, Kaufmann SH. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc Natl Acad Sci U S A. 2011 Feb 22; 108(8):3406–3411.
- Kaufman B, Shapira-Frommer R, Schmutzler RK, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015 Jan 20;33(3):244–250.
- Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012 Apr 12;366(15):1382–1392.
- Oza AM, Cibula D, Benzaquen AO, et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: a randomised phase 2 trial. Lancet Oncol. 2015 Jan;16(1):87–97.
- Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014 Jul;15(8):852–861.
- Van Der Noll R, Marchetti S, Steeghs N, et al. Long-term safety and anti-tumour activity of olaparib monotherapy after combination with carboplatin and paclitaxel in patients with advanced breast, ovarian or fallopian tube cancer. Br J Cancer. 2015 Jul 28;113(3):396–402.
- Domchek SM, Aghajanian C, Shapira-Frommer R, et al. Efficacy and safety of olaparib monotherapy in germline BRCA1/2 mutation carriers with advanced ovarian cancer and three or more lines of prior therapy. Gynecol Oncol. 2016 Feb;140(2):199–203.
- Lee JM, Hays JL, Annunziata CM, et al. Phase I/Ib study of olaparib and carboplatin in BRCA1 or BRCA2 mutation-associated breast or ovarian cancer with biomarker analyses. J Natl Cancer Inst. 2014 May 19;106(6):dju089.
- Bindra RS, Schaffer PJ, Meng A, et al. Down-regulation of Rad51 and decreased homologous recombination in hypoxic cancer cells. Mol Cell Biol. 2004 Oct;24(19):8504–8518.
- Lim JJ, Yang K, Taylor-Harding B, et al. VEGFR3 inhibition chemosensitizes ovarian cancer stemlike cells through down-regulation of BRCA1 and BRCA2. Neoplasia. 2014 Apr;16(4):343,53.e1-2.
- Liu JF, Barry WT, Birrer M, et al. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: a randomised phase 2 study. Lancet Oncol. 2014 Oct;15(11):1207–1214.
- Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006 Aug;7(8):606–619.
- Kumar A, Fernandez-Capetillo O, Carrera AC. Nuclear phosphoinositide 3-kinase beta controls double-strand break DNA repair. Proc Natl Acad Sci U S A. 2010 Apr 20;107(16):7491–7496.
- Juvekar A, Burga LN, Hu H, et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov. 2012 Nov;2(11):1048–1063.
- Ibrahim YH, Garcia-Garcia C, Serra V, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012 Nov;2(11):1036–1047.
- Gonzalez-Billalabeitia E, Seitzer N, Song SJ, et al. Vulnerabilities of PTEN-TP53-deficient prostate cancers to compound PARP-PI3K inhibition. Cancer Discov. 2014 Aug;4(8):896–904.
- Johnson N, Li YC, Walton ZE, et al. Compromised CDK1 activity sensitizes BRCA-proficient cancers to PARP inhibition. Nat Med. 2011 Jun 26;17(7):875–882.
- Johnson SF, Cruz C, Greifenberg AK, et al. CDK12 inhibition reverses de novo and acquired PARP inhibitor resistance in BRCA wild-type and mutated models of triple-negative breast cancer. Cell Rep. 2016 Nov 22;17(9):2367–2381.
- Jirawatnotai S, Hu Y, Michowski W, et al. A function for cyclin D1 in DNA repair uncovered by protein interactome analyses in human cancers. Nature. 2011 Jun 8;474(7350):230–234.
- Bergs JW, Krawczyk PM, Borovski T, et al. Inhibition of homologous recombination by hyperthermia shunts early double strand break repair to non-homologous end-joining. DNA Repair (Amst). 2013 Jan 1;12(1):38–45.
- Krawczyk PM, Eppink B, Essers J, et al. Mild hyperthermia inhibits homologous recombination, induces BRCA2 degradation, and sensitizes cancer cells to poly (ADP-ribose) polymerase-1 inhibition. Proc Natl Acad Sci U S A. 2011 Jun 14;108(24):9851–9856.
- Sakai W, Swisher EM, Karlan BY, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008 Feb 28;451(7182):1116–1120.
- Swisher EM, Sakai W, Karlan BY, et al. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008 Apr 15; 68(8):2581–2586.
- Easton DF, Deffenbaugh AM, Pruss D, et al. A systematic genetic assessment of 1,433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am J Hum Genet. 2007 Nov;81(5):873–883.
- Ter Brugge P, Kristel P, Van Der Burg E, et al. Mechanisms of therapy resistance in patient-derived xenograft models of BRCA1-deficient breast cancer. J Natl Cancer Inst. 2016 Jul 5;108(11). Print 2016 Nov. doi:10.1093/jnci/djw148.
- Patch AM, Christie EL, Etemadmoghadam D, et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015 May 28;521(7553):489–494.
- Cao L, Xu X, Bunting SF, et al. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol Cell. 2009 Aug 28;35(4):534–541.
- Iwabuchi K, Bartel PL, Li B, et al. Two cellular proteins that bind to wild-type but not mutant p53. Proc Natl Acad Sci U S A. 1994 Jun 21;91(13):6098–6102.
- Wang B, Matsuoka S, Carpenter PB, et al. 53BP1, a mediator of the DNA damage checkpoint. Science. 2002 Nov 15;298(5597):1435–1438.
- Bouwman P, Aly A, Escandell JM, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010 Jun;17(6):688–695.
- Bunting SF, Callen E, Wong N, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010 Apr 16;141(2):243–254.
- Kass EM, Moynahan ME, Jasin M. Loss of 53BP1 is a gain for BRCA1 mutant cells. Cancer Cell. 2010 May 18;17(5):423–425.
- Chapman JR, Barral P, Vannier JB, et al. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell. 2013 Mar 7;49(5):858–871.
- Gottipati P, Vischioni B, Schultz N, et al. Poly(ADP-ribose) polymerase is hyperactivated in homologous recombination-defective cells. Cancer Res. 2010 Jul 1;70(13):5389–5398.
- Liu X, Han EK, Anderson M, et al. Acquired resistance to combination treatment with temozolomide and ABT-888 is mediated by both base excision repair and homologous recombination DNA repair pathways. Mol Cancer Res. 2009 Oct;7(10):1686–1692.
- Zaremba T, Ketzer P, Cole M, et al. Poly(ADP-ribose) polymerase-1 polymorphisms, expression and activity in selected human tumour cell lines. Br J Cancer. 2009 Jul 21;101(2):256–262.
- McCabe N, Turner NC, Lord CJ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006 Aug 15;66(16):8109–8115.
- Moghadasi S, Hofland N, Wouts JN, et al. Variants of uncertain significance in BRCA1 and BRCA2 assessment of in silico analysis and a proposal for communication in genetic counselling. J Med Genet. 2013 Feb;50(2):74–79.
- Bouwman P, Van Der Gulden H, Van Der Heijden I, et al. A high-throughput functional complementation assay for classification of BRCA1 missense variants. Cancer Discov. 2013 Oct;3(10):1142–1155.
- Eccles DM, Mitchell G, Monteiro AN, et al. BRCA1 and BRCA2 genetic testing-pitfalls and recommendations for managing variants of uncertain clinical significance. Ann Oncol. 2015 Oct;26(10):2057–2065.
- Norquist B, Wurz KA, Pennil CC, et al. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol. 2011 Aug 1;29(22):3008–3015.
- Joosse SA, van Beers EH, Tielen IH, et al. Prediction of BRCA1-association in hereditary non-BRCA1/2 breast carcinomas with array-CGH. Breast Cancer Res Treat. 2009 Aug;116(3):479–489.
- Vollebergh MA, Lips EH, Nederlof PM, et al. An aCGH classifier derived from BRCA1-mutated breast cancer and benefit of high-dose platinum-based chemotherapy in HER2-negative breast cancer patients. Ann Oncol. 2011 Jul;22(7):1561–1570.
- Davies H, Glodzik D, Morganella S, et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med. 2017 Apr;23(4):517–525.
- Norquist BM, Harrell MI, Brady MF, et al. Inherited mutations in women with ovarian carcinoma. JAMA Oncol. 2016 Apr;2(4):482–490.
- Watkins JA, Irshad S, Grigoriadis A, et al. Genomic scars as biomarkers of homologous recombination deficiency and drug response in breast and ovarian cancers. Breast Cancer Res. 2014 Jun 3;16(3):211.
- Timms KM, Abkevich V, Hughes E, et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014 Dec 5;16(6):475,014-0475-x.
- Patel J, Sehouli J, Timms K, et al. Characteristics of homologous recombination deficiency (HRD) in paired primary and recurrent high-grade serous ovarian cancer (HGSOC). Ann Oncol. 2016;27(suppl_6):113
- Telli ML, Timms KM, Reid J, et al. Homologous recombination deficiency (HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer. Clin Cancer Res. 2016 Aug 1;22(15):3764–3773.
- Haaf T, Golub EI, Reddy G, et al. Nuclear foci of mammalian Rad51 recombination protein in somatic cells after DNA damage and its localization in synaptonemal complexes. Proc Natl Acad Sci U S A. 1995 Mar 14;92(6):2298–2302.
- Yuan SS, Lee SY, Chen G, et al. BRCA2 is required for ionizing radiation-induced assembly of Rad51 complex in vivo. Cancer Res. 1999 Aug 1;59(15):3547–3551.
- Shah MM, Dobbin ZC, Nowsheen S, et al. An ex vivo assay of XRT-induced Rad51 foci formation predicts response to PARP-inhibition in ovarian cancer. Gynecol Oncol. 2014 Aug;134(2):331–337.
- Mukhopadhyay A, Elattar A, Cerbinskaite A, et al. Development of a functional assay for homologous recombination status in primary cultures of epithelial ovarian tumor and correlation with sensitivity to poly(ADP-ribose) polymerase inhibitors. Clin Cancer Res. 2010 Apr 15;16(8):2344–2351.
- van Veelen LR, Essers J, van de Rakt MW, et al. Ionizing radiation-induced foci formation of mammalian Rad51 and Rad54 depends on the Rad51 paralogs, but not on Rad52. Mutat Res. 2005 Jul 1;574(1–2):34–49.
- Wohlschlegel JA, Kutok JL, Weng AP, et al. Expression of geminin as a marker of cell proliferation in normal tissues and malignancies. Am J Pathol. 2002 Jul;161(1):267–273.
- Naipal KA, Verkaik NS, Ameziane N, et al. Functional ex vivo assay to select homologous recombination-deficient breast tumors for PARP inhibitor treatment. Clin Cancer Res. 2014 Sep 15;20(18):4816–4826.
- Mutter RW, Riaz N, Ng CK, et al. Bi-allelic alterations in DNA repair genes underpin homologous recombination DNA repair defects in breast cancer. J Pathol. 2017 Mar 15. doi:10.1002/path.4890.
- Graeser M, McCarthy A, Lord CJ, et al. A marker of homologous recombination predicts pathologic complete response to neoadjuvant chemotherapy in primary breast cancer. Clin Cancer Res. 2010 Dec 15;16(24):6159–6168.