
ABSTRACT
Despite the significant efforts devoted to decipher the particular protein features that encode for a prion or prion-like behavior, they are still poorly understood. The well-characterized yeast prions constitute an ideal model system to address this question, because, in these proteins, the prion activity can be univocally assigned to a specific region of their sequence, known as the prion forming domain (PFD). These PFDs are intrinsically disordered, relatively long and, in many cases, of low complexity, being enriched in glutamine/asparagine residues. Computational analyses have identified a significant number of proteins having similar domains in the human proteome. The compositional bias of these regions plays an important role in the transition of the prions to the amyloid state. However, it is difficult to explain how composition alone can account for the formation of specific contacts that position correctly PFDs and provide the enthalpic force to compensate for the large entropic cost of immobilizing these domains in the initial assemblies. We have hypothesized that short, sequence-specific, amyloid cores embedded in PFDs can perform these functions and, accordingly, act as preferential nucleation centers in both spontaneous and seeded aggregation. We have shown that the implementation of this concept in a prediction algorithm allows to score the prion propensities of putative PFDs with high accuracy. Recently, we have provided experimental evidence for the existence of such amyloid cores in the PFDs of Sup35, Ure2, Swi1, and Mot3 yeast prions. The fibrils formed by these short stretches may recognize and promote the aggregation of the complete proteins inside cells, being thus a promising tool for targeted protein inactivation.
Protein misfolding and aggregation is associated with a broad range of human disorders, including Parkinson's and Alzheimer's diseases.Citation1 The common underlying cause behind these pathologies is the conversion of specific soluble proteins into insoluble and highly ordered fibrillar aggregates, collectively referred as amyloid fibrils. Analysis by X-ray diffraction of these fibrils indicates that, in most cases, the polypeptide chains are embedded in an extended cross-β-conformation running perpendicular to the fibril long axis.Citation2 The folding of globular proteins into native structures relies on the establishment of an extensive network of interactions involving most of the protein sequence.Citation3 In contrast, amyloid self-assembly seems to obey, in many cases, the “short-stretch hypothesis,” according to which, the formation of this supramolecular structure is first nucleated by the intermolecular contacts formed by a reduced number of specific short regions in the protein, named aggregation hot-spots (HS) or short aggregation-prone regions (APRs)Citation4 (). These stretches are generally around 5–10 residues in length, with a predominant hydrophobic character and a low net charge.Citation5
Figure 1. Scheme of the initial intermolecular contacts leading to amyloid formation according to different models. (A) The amyloid classical model relies on the initial establishment of interactions (thin lines) between short APRs of about 5–10 residues with a predominant hydrophobic character (red boxes). (B) The prion compositional model relies on the initial establishment of a large number of diffuse weak interactions (thin lines) along the PFD. (C) In the new model for prion and prion-like proteins, the first contacts (thin lines) are formed between short amyloidogenic regions (red boxes) embedded into a Q/N-rich disordered region. The amyloid core is longer than those in the classical amyloid model, and, due to their particular composition, display less amyloidogenic potential. This allows the protein to remain soluble until needed and results in assemblies with a significant degree of brittleness.
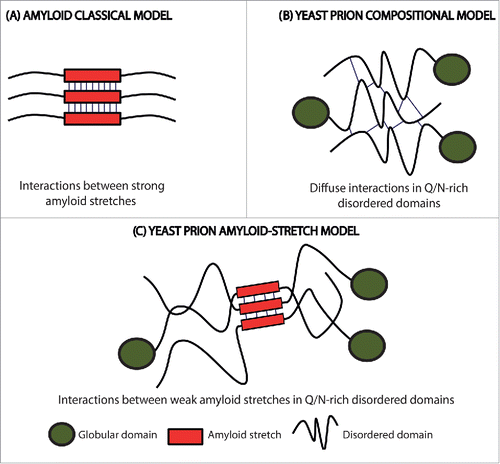
Prions are considered a subclass of amyloids with the ability to propagate their aggregated conformation and thus, to become potentially infectious. The prion phenomenon is best known by its association with spongiform encephalopathies in mammals,Citation6 but increasing evidence indicates that similar templating mechanisms are exploited by nature for a variety of functional purposes,Citation7 being protein-based epigenetic inheritance in yeast the best characterized of these processes.Citation8,9 Like their mammalian counterparts, yeast prions undergo a self-perpetuating conversion into an amyloid conformation that shift the function of these proteins, which, in the case of yeast, might promote the expression of novel and, eventually, beneficial phenotypic traits.Citation10 A common feature of many of these yeast prions is the presence of a long and intrinsically disordered region, which is enriched in glutamine/asparagine (Q/N) residues and usually depleted in hydrophobic ones, named prion-forming domain (PFD). This domain is both sufficient and necessary for prion conversion.Citation11 However, it should be mentioned that the presence of a low complexity Q/N-rich region is not a mandatory requirement for prion formation, since an increasing number of polypeptides able to promote protein-based inheritance are being discovered in fungi and other organisms that lack this specific compositional signature. What it seems to be common to all PFDs is their intrinsically disordered nature in the soluble state of the prion protein.Citation12
Whereas almost any protein bears the potential to form amyloid structures,Citation13 only a small set of amyloid assemblies shows prion behavior in natural environments. Indeed, despite the structural similarity between the aggregated states of amyloids and prions, classical amyloid prediction algorithms fail to identify prion propensities in protein sequences.Citation4 Actually, in spite of the particular sequential features of Q/N-rich PFDs being known since long time ago, we have little information on how they encode for a prion-like behavior. Traditionally, the self-assembly of these polypeptides have been thought to ultimately depend on their particular amino acid composition, with amyloid formation relying on the establishment of a large number of weak interactions distributed over the long PFDCitation14 (). A number of prion protein prediction programs have been developed according to this rational. These algorithms score protein sequences according to their compositional similarity with the PFDs of a few, well-characterized, yeast prions. They have allowed to search for new PFDs candidates in previously unexplored proteomes.Citation15 These predicted regions have been named prion-like domains (PrLDs), and the proteins holding them, prion-like proteins. Interestingly enough, the analysis of the human proteome revealed that a significant number of proteins displaying PrLDs are enriched in DNA or RNA binding domains and involved in regulatory functions, with some of them being associated with neurodegenerative diseases.Citation16 Examples of these algorithms are DIANA,Citation17 LPSs,Citation18 PAPA,Citation19 PrionScan,Citation20,21 and PLAAC.Citation22
The success of composition-based predictors in the identification of new yeast proteins behaving like bona fide prions,Citation15 together with the failure of classical amyloid prediction algorithms to do this task, has led to assume that the “short-stretch hypothesis” does not apply for prion or prion-like proteins.Citation11 This assumption was apparently consistent with the observation that, due to their disordered nature, PFDs are devoid of high local concentrations of hydrophobic residues.
The excellent hydrogen bonding capability of Q and N residuesCitation16 has led to propose that the formation of a diffuse network of hydrogen bonds would initiate the self-assembly reaction in PFDs. However, because hydrogen bonds between these residues are only slightly more stable that the ones they establish with the solvent, it is difficult to envision how the initial formation of delocalized and globally weak contacts can compensate enthalpically for the high entropic cost of immobilizing a long disordered and presumably highly flexible prion domain. Therefore, we decided to re-explore the possibility that the presence of short stretches with a significant amyloid propensity embedded in the disordered Q/N-rich regions of PFDs might act as nucleating regions for amyloid assembly.Citation17 Two reasons argued that the amyloid potential of these APRs, if they exist, should be weaker than those found in disease-linked amyloid polypeptides. First, despite PFDs should keep a certain amyloid capacity that permits the switch toward the insoluble state, their physicochemical characteristics should be compatible with the protein being soluble under physiological conditions, at least during part of its lifetime. Fully exposed, highly aggregation-prone stretches will shift irreversibly the equilibrium toward the insoluble state, precluding any functional transition. Second, the amyloids formed by yeast prions should display a certain degree of brittleness (), since this feature is crucial for their chaperone-assisted fragmentation and subsequent propagation between mother and daughter cells.Citation18 The presence of a very strong amyloid core would prevent, or at least decrease, the fragmentation rate and thus reduce prion propagation potential.
Amyloid stretches in PFDs should fulfill two apparently contradictory properties: allow the domain to remain soluble and disordered and provide the nucleation force for amyloid formation. It is known, that polar and charged residues favor disorder, whereas hydrophobic amino acids favor aggregation. Then, how these 2 properties can be encoded at the same time in a short sequence stretch? By computationally analyzing the ability of the 20 proteinogenic amino acids to promote disorder and aggregation, we discovered that N and Q are the residues that best balance amyloid and disorder propensities.Citation11 We proposed that this unique property accounts for the over-representation of these residues in yeast PFDs.Citation11 Tyrosine (Y) is the most abundant hydrophobic residue in yeast PFDs.Citation15 Our analysis indicated that Y is clearly superior to the rest of apolar residues in terms of disorder propensity, appearing thus as the best residue to endorse prionogenic Q/N-rich amyloid cores with increased amyloid potential without disturbing significantly the PFDs disorder properties. However, computational simulations soon demonstrated that a 5–10 residues long stretch based on Q/N residues would not have enough amyloid potential to drive by itself the transition toward the fibrillar state, even if it contains a certain number of Y residues. Thus, we hypothesized that the amyloid cores of PFDs should be longer, in such a way that the amyloid potential would be more distributed than in classical amyloid stretches; each residue having an average lower contribution, but with more residues contributing to assembling driving force. We implemented this notion in a novel PFDs prediction algorithm named pWALTZ.Citation17 pWALTZ predicts the 21 residues long sequence stretch with the average highest amyloidogenic potential into a Q/N-rich context and classifies proteins into the prion/non-prion categories according to the presence and the potency of these stretches. Interestingly enough, this approach displays better accuracy in discriminating prion propensity than algorithms relying only in the compositional model.Citation17 Later on, this algorithm was incorporated into the publically accessible PrionW webserver.Citation19
The fact that an algorithm based on the “short-stretch amyloid hypothesis” can infer the prion propensity of a sequence suggests, but does not prove, that these stretches really exist. Recently, we have provided experimental evidence for the presence of short amyloid cores in the PFDs of some of the most representative canonical yeast prions: Sup35, Ure2, Swi1 and Mot3, which are all implicated in regulatory functions relevant for cell adaptation to the changing environment. Sup35 is a component of translation termination complex. When Sup35 is converted to its prion form [PSI+], stop codons can be read-through increasing the phenotypic variability and therefore multiplying the chances to attain optimal yeast fitness.Citation20 Ure2 is a negative regulator of enzymes involved in nitrogen metabolism. In its prion form, named as [URE3], the repression is removed and the yeast cell can import ureidosuccinate.Citation21 Swi1, a subunit of the SWI/SNF ATP-dependent chromatin-remodeling complex implicated in the expression of 6% of the yeast genome, can become a prion named [SWI+]. Cells containing the prion [SWI+] show a phenotype indicative of partial loss of function of SWI/SNF22. Mot3 transcriptional factor in its prion form [MOT3+] regulates the facultative acquisition of multicellular structures in response to natural environmental signals.Citation23
We first searched for the presence of amyloid cores into the PFDs of the above described 4 proteins using the pWALTZ formalism. In all the cases, we could identify at least one sequence that fulfills the algorithm requirements.Citation24 As expected, classical aggregation predictors like TANGO,Citation25 AGGRESCANCitation26 or PASTACitation27 were unable to identify these amyloid stretches, since the abundance of Q/N residues in these sequences is interpreted by these programs as a signature of low amyloid potential. This highlights how, as hypothesized, despite the “short-stretch amyloid hypothesis” would apply for both classical amyloids and prions, the specific features of their cores differ significantly.
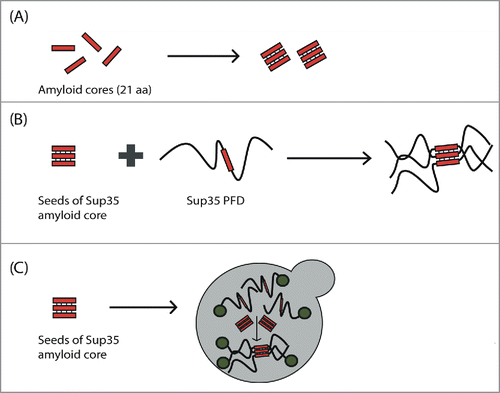
We synthesized the peptides corresponding to the 4 predicted cores and show that all of them were able to experiment a transition between an initially disordered and soluble conformation and a β-sheet rich state, as determined by Far-UV circular dichroism and infrared spectroscopy. Moreover, analysis by Thioflavin T binding, transmission electron microscopy and X-ray diffraction, all converged to indicate that these β-sheet rich conformations correspond to archetypical amyloid fibrils exhibiting a cross-β structure (). The fibrils formed by these amyloid cores were able to seed the aggregation of their corresponding soluble form, but, despite all them share a similar composition, with ∼1/3 of their residues being Q/N, no cross-seeding effect between cores could be observed. This indicated that, at least for these particular PFDs segments, they were sequence-specific contacts that drove their aggregation. Importantly, in the case of Sup35, the fibrils formed by the peptide were able to promote the accelerated aggregation of the complete PFD in vitro ().Citation24 Indeed, they seeded the reaction with higher efficiency than the fibrils formed by the entire PFD, which strongly argues that this stretch was able to nucleate the reaction by recognizing the homologous sequence in the soluble and disordered PFD. This will explain why, when the fibrils formed by the amyloid core of Sup35 are introduced in living yeast cells, they are able to promote the shift of a fraction of the population to the prionic state (),Citation24 behaving thus as autonomous propagation-competent entities. This suggests that they identified regions may act as key regulators of the phenotypic conversion induced by yeast prions. Chaperone-mediated fragmentation is indispensable to propagate yeast prions; despite speculative, it is tempting to propose that, upon chaperone cleavage, fragments containing the identified amyloid cores might act as preferential seeds in the conversion of the soluble prion proteins present in daughter cells toward the amyloid state.
Figure 2. Scheme of the experimental evidence for the presence of amyloid cores in canonical yeast prions. (A) Amyloid cores from Sup35, Ure2, Swi1 and Mot3 are able to make the transition from the soluble and disordered state to amyloid fibrils with cross-β structure. (B) The Sup35 amyloid core is able to promote (seed) the aggregation of the complete Sup35 PFD in vitro. (C) Introduction of Sup35 amyloid core seeds into yeast is able to induce the aggregation of the endogenous Sup35 protein in vivo and, consequently, the expression of the prionic phenotype.
Overall, our results indicate that the sustained mechanism of functional prion assembly would resemble more than initially thought to that of disease-linked amyloids. This view is not in opposition to the previous compositional model, but it goes a step further by defining a region with a higher probability to initiate the conformational conversion. The Q/N compositional bias would define the structurally disordered context, while providing a cryptic and distributed aggregation propensity that can zipper the complete PFD upon amyloid core positioning and subsequent nucleation. In a way, the reaction would resemble that occurring in polyQ diseases.Citation28 In these disorders, the expansion of an intrinsically disordered polyQ tract flanking a folded globular domain results in protein aggregation. Despite the expanded polyQ stretch is critical for amyloid assembly, it is clear that its lack of sequence specificity makes difficult that it could encode an accurate, in register, disposition of the polypeptide chains at the beginning of the aggregation reaction. Accordingly, increasing evidence indicates that, in these proteins, aggregation is initiated by contacts between specific APRs in, or close to, the globular domain and only latter it is propagated to the polyQ segment.Citation29,30 This sequence-specific initiation of amyloid formation would allow a proper pairing and hydrogen bonding of the residues in the monotonic polyQ track. In a similar manner, in addition to contribute to the initial force for aggregation, amyloid cores in PFDs would allow an accurate positioning and intermolecular interaction of the disordered Q/N-rich regions that flank them. The same scheme will apply for seeding reactions, making cross-seeding between different PFDs less probable, despite they share very similar composition.
We want to make clear that the selected size of 21 residues for amyloid cores is arbitrary. It was the length that performed best in discriminating prion versus non prion behavior in Q/N-rich domainsCitation17 and also because this length has been shown to coincide with the minimal motif accounting for HET-S prion transmission.Citation31 However, we assume that amyloid cores of different lengths would indeed exist, the size depending on both their composition and the sequential context in which they are embedded. In addition, we do not propose that the identified amyloid cores are the exclusive players in the aggregation process of the studied PFDs, they can clearly exist other short regions that collaborate either in a cooperative or additive way to the process. This is clearly the case of Sup35, for which a region adjacent to the N-terminus of the assayed amyloid-core has been shown to play an essential role in the aggregation and propagation of this protein.Citation32 In this sense, we are developing a version of pWALTZ able to identify and compare all the putative amyloid cores in a Q/N-rich sequence. The preliminary analysis indicates that the average number of amyloid cores in PFDs is rather small.
It is interesting to notice that the characteristics of amyloid cores in PFDs resemble that of Short Linear Motifs (SLiMs)Citation33 found in intrinsically disordered proteins. SLiMs are short stretches of adjacent amino acids that mediate protein-protein association, also known as linear motifs (LMs) and molecular recognition features (MoRFs). SLiMs are usually constituted by a short stretch of contiguous amino acids, and their binding can be modulated by residues outside this region. As it happens with yeast prion amyloid cores, these SLiMs are generally placed in unstructured regions and, when their binding partners are absent, they lack stable tertiary structure.Citation34 It has been proposed that the short length and the small number of essential residues defining these SLiMs make them much more easy to evolve through point mutations than equivalent structural binding motifs in the context of globular domains.Citation34 The new rudimentary motif created by these point mutations can be consequently selected positively or negatively by evolutionary pressures, to produce a functional SLiM or to eliminate a pernicious interaction, respectively.Citation34 It is feasible that a similar mechanism might apply to the amyloid cores we have identified in yeast prions. Genetic mutations generating new amyloid cores would be selected as long as they result advantageous for the cell and/or the population fitness. From an evolutionary point of view, the fact that sequences fully exposed to solvent and with a significant amyloid potential, as those we have identified in PFDs, have not been purged out by natural selection can only be explained if they serve for functional purposes,Citation35 since there is a strong selective pressure to reduce the amyloidogenic potential of protein sequences.Citation36 The presence of these regions is inherently risky, since mutations that increase their amyloid potential can shift the equilibrium to an aggregated and potentially toxic state. This is exactly what happens in the case of human hnRNPA1 and hnRNA2, two prion-like RNA-binding proteins which genetic mutation is associated with multisystem proteinopathy and amyotrophic lateral sclerosis.Citation37 The analysis of these pathogenic mutations with pWALTZ indicated that they map at the predicted amyloid cores, increasing their amyloid propensity.Citation17 This mutation-induced pro-aggregational effect might also occur in other human prion-like proteins and explain why, as a group, they appear to be linked to neurological disease.Citation38 pWALTZ might turn to be useful in the detection and prediction of pathogenic mutations in this sub-proteome.
The ability of Sup35 amyloid core to fibrillate and to induce the intracellular aggregation of the homologous yeast prion protein, suggests that a similar strategy can be used for the targeted inactivation of human prion-like proteins. A related approach has been recently shown to be effective to target the vascular endothelial growth factor 2 (VEGFR2), a protein containing amyloidogenic segments, but which is not known to aggregate in either pathological or normal conditions. Citation39 The internalization inside cells of a synthetic peptide, corresponding to a tandem repeat of a short amyloidogenic region of VEGFR2, induced its aggregation in vivo through a direct amyloid interaction, inactivating the protein and, accordingly, inhibiting VEGFR2-dependent tumor growth in a mouse tumor model. Interestingly enough, despite VEGFR2 exhibited a significant number of amyloid stretches, those belonging to the signal peptide exhibited the best performance. This is likely because in contrast to the ones mapping in the globular domain, they are already exposed to solvent, before the signal peptide is processed. Computational predictions indicate that amyloid cores in human PrLDs are placed in a similar structural context, which anticipates that they can be potentially targeted by homologous fibrillated peptides to inactivate the function of selected prion-like proteins. Work in this direction is ongoing in our lab.
ABBREVIATIONS
| APRs | = | short aggregation-prone regions |
| PFD | = | prion-forming domain |
| PrLD | = | prion-like domain |
| SLiM | = | short linear motifs |
| VEGFR2 | = | vascular endothelial growth factor 2 |
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
FUNDING
This work was supported by the Spanish “Ministerio de Economia y Competitividad” [BFU2013–44763-P] to S.V and by ICREA, ICREA-Academia 2015 to S.V.
REFERENCES
- Invernizzi G, Papaleo E, Sabate R, Ventura S. Protein aggregation: Mechanisms and functional consequences. Int J Biochem Cell Biol 2012; 44(9):1541-54; PMID:22713792; http://dx.doi.org/10.1016/j.biocel.2012.05.023
- Sabaté R, Ventura S. Cross-β-sheet supersecondary structure in amyloid folds: techniques for detection and characterization. Methods Mol Biol 2012; 932:237-57.
- Anfinsen CB. Principles that govern the folding of protein chains. Science 1973; 181(4096):223-30; PMID:4124164; http://dx.doi.org/10.1126/science.181.4096.223
- Toombs JA, Petri M, Paul KR, Kan GY, Ben-Hur A, Ross ED. De novo design of synthetic prion domains. Proc Natl Acad Sci U S A 2012; 109(17):6519-24; PMID:22474356; http://dx.doi.org/10.1073/pnas.1119366109
- de Groot NS, Pallarés I, Avilés FX, Vendrell J, Ventura S. Prediction of “hot spots” of aggregation in disease-linked polypeptides. BMC Struct Biol 2005; 5:1-15; PMID:15663787; http://dx.doi.org/10.1186/1472-6807-5-18
- Collins SJ, Lawson VA, Masters CL. Transmissible spongiform encephalopathies. Lancet 2004; 363(9402):51-61; PMID:14723996; http://dx.doi.org/10.1016/S0140-6736(03)15171-9
- Si K. Prions: What are they good for? Annu Rev Cell Dev Biol 2015; 31(1):149-69.
- Chien P, Weissman JS, DePace AH. Emerging principles of conformation-based prion inheritance. Annu Rev Biochem 2004; 73(1):617-56; PMID:15189155; http://dx.doi.org/10.1146/annurev.biochem.72.121801.161837
- Liebman SW, Chernoff YO. Prions in yeast. Genetics 2012; 191(4):1041-72; PMID:22879407; http://dx.doi.org/10.1534/genetics.111.137760
- Wickner RB, Edskes HK, Gorkovskiy A, Bezsonov EE, Stroobant EE. Yeast and fungal prions: amyloid-handling systems, amyloid structure, and prion biology. Adv Genet 2016; 93:191-236; PMID:26915272
- Sabate R, Rousseau F, Schymkowitz J, Batlle C, Ventura S. Amyloids or Prions? That is the Question. Prion 2015; 9:200-6; PMID:26039159; http://dx.doi.org/10.1080/19336896.2015.1053685
- Chakrabortee S, Byers JS, Jones S, Garcia DM, Bhullar B, Chang A, She R, Lee L, Fremin B, Lindquist S, et al. Intrinsically disordered proteins drive emergence and inheritance of biological traits. Cell 2016; 167(2):369-81; PMID:27693355; http://dx.doi.org/10.1016/j.cell.2016.09.017
- Chiti F, Webster P, Taddei N, Clark A, Stefani M, Ramponi G, Dobson CM. Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc Natl Acad Sci U S A 1999; 96(7):3590-4; PMID:10097081; http://dx.doi.org/10.1073/pnas.96.7.3590
- Ross ED, Edskes HK, Terry MJ, Wickner RB. Primary sequence independence for prion formation. Proc Natl Acad Sci 2005; 102(36):12825-30; PMID:16123127; http://dx.doi.org/10.1073/pnas.0506136102
- Alberti S, Halfmann R, King O, Kapila A, Lindquist S. A Systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 2009; 137(1):146-58; PMID:19345193; http://dx.doi.org/10.1016/j.cell.2009.02.044
- Stapley BJ, Doig AJ. Hydrogen bonding interactions between glutamine and asparagine in α-helical peptides. J Mol Biol 1997; 272(3):465-73; PMID:9325104; http://dx.doi.org/10.1006/jmbi.1997.1262
- Sabate R, Rousseau F, Schymkowitz J, Ventura S. What makes a protein sequence a prion? PLoS Comput Biol 2015; 11(1):e1004013; PMID:25569335; http://dx.doi.org/10.1371/journal.pcbi.1004013
- Tanaka M, Collins SR, Toyama BH, Weissman JS. The physical basis of how prion conformations determine strain phenotypes. Nature 2006; 442(7102):585-9; PMID:16810177; http://dx.doi.org/10.1038/nature04922
- Zambrano R, Conchillo-Sole O, Iglesias V, Illa R, Rousseau F, Schymkowitz J, Sabate R, Daura X, Ventura S. PrionW: a server to identify proteins containing glutamine/asparagine rich prion-like domains and their amyloid cores. Nucleic Acids Res 2015; 43(W1):W331-7; PMID:25977297; http://dx.doi.org/10.1093/nar/gkv490
- Serio TR, Lindquist. The yeast prion [PSI+]: molecular insights and functional consequences. Adv Protein Chem 2002; 59:391-412.
- Wickner RB. [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science 1994; 264:5669; PMID:7909170; http://dx.doi.org/10.1126/science.7909170
- Du Z, Park KW, Yu H, Fan Q, Li L. Newly identified prion linked to the chromatin-remodeling factor Swi1 in Saccharomyces cerevisiae. Nat Genet 2008; 40(4):460-5; PMID:18362884; http://dx.doi.org/10.1038/ng.112
- Holmes DL, Lancaster AK, Lindquist S, Halfmann R. Heritable Remodeling of Yeast Multicellularity by an Environmentally Responsive Prion. Cell 2013; 153(1):153-65; PMID:23540696; http://dx.doi.org/10.1016/j.cell.2013.02.026
- Sant'Anna R, Fernández MR, Batlle C, Navarro S, de Groot NS, Serpell L, Ventura S. Characterization of amyloid cores in prion domains. Sci Rep 2016; 6:34274; PMID:27686217; http://dx.doi.org/10.1038/srep34274
- Fernandez-Escamilla A-M, Rousseau F, Schymkowitz J, Serrano L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol 2004; 22(10):1302-6; PMID:15361882; http://dx.doi.org/10.1038/nbt1012
- de Groot NS, Castillo V, Graña-Montes R, Ventura S. AGGRESCAN: method, application, and perspectives for drug design. Methods Mol Biol 2012; 819:199-220; PMID:22183539
- Walsh I, Seno F, Tosatto SCE, Trovato A. PASTA 2.0: an improved server for protein aggregation prediction. Nucleic Acids Res 2014; 42:W301-7; PMID:24848016; http://dx.doi.org/10.1093/nar/gku399
- Petrakis S, Schaefer MH, Wanker EE, Andrade-Navarro MA. Aggregation of polyQ-extended proteins is promoted by interaction with their natural coiled-coil partners. Bioessays 2013; 35(6):503-7; PMID:23483542; http://dx.doi.org/10.1002/bies.201300001
- Lupton CJ, Steer DL, Wintrode PL, Bottomley SP, Hughes VA, Ellisdon AM. Enhanced Molecular Mobility of Ordinarily Structured Regions Drives Polyglutamine Disease. J Biol Chem 2015; 290(40):24190-200; PMID:26260925; http://dx.doi.org/10.1074/jbc.M115.659532
- Scarff CA, Almeida B, Fraga J, Macedo-Ribeiro S, Radford SE, Ashcroft AE. Examination of Ataxin-3 (atx-3) Aggregation by Structural Mass Spectrometry Techniques: A Rationale for Expedited Aggregation upon Polyglutamine (polyQ) Expansion. Mol Cell Proteomics 2015; 14(5):1241-53; PMID:25700012; http://dx.doi.org/10.1074/mcp.M114.044610
- Wan W, Stubbs G. Fungal prion HET-s as a model for structural complexity and self-propagation in prions. Proc Natl Acad Sci U S A 2014; 111(14):5201-6; PMID:24706820; http://dx.doi.org/10.1073/pnas.1322933111
- Osherovich LZ, Cox BS, Tuite MF, Weissman JS. Dissection and design of yeast prions. PLoS Biol 2004; 2(4):e86; http://dx.doi.org/10.1371/journal.pbio.0020086
- Mészáros B, Dosztányi Z, Simon I, Blow N, Jones S, Thornton J, Diella F, Haslam N, Chica C, Budd A, et al. Disordered binding regions and linear motifs—bridging the gap between two models of molecular recognition. PLoS One 2012; 7(10):e46829; PMID:23056474; http://dx.doi.org/10.1371/journal.pone.0046829
- Van Roey K, Uyar B, Weatheritt RJ, Dinkel H, Seiler M, Budd A, Gibson TJ, Davey NE. Short linear motifs: ubiquitous and functionally diverse protein interaction modules directing cell regulation. Chem Rev 2014; 114(13):6733-78; PMID:24926813; http://dx.doi.org/10.1021/cr400585q
- Malinovska L, Kroschwald S, Alberti S. Protein disorder, prion propensities, and self-organizing macromolecular collectives. Biochim Biophys Acta 2013; 1834(5):918-31; PMID:23328411; http://dx.doi.org/10.1016/j.bbapap.2013.01.003
- Buck PM, Kumar S, Singh SK. On the role of aggregation prone regions in protein evolution, stability, and enzymatic catalysis: insights from diverse analyses. PLoS Comput Biol 2013; 9(10):e1003291; PMID:24146608; http://dx.doi.org/10.1371/journal.pcbi.1003291
- Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013; 495(7442):467-73; PMID:23455423; http://dx.doi.org/10.1038/nature11922
- An L, Harrison PM. The evolutionary scope and neurological disease linkage of yeast-prion-like proteins in humans. Biol Direct 2016; 11:32; PMID:27457357; http://dx.doi.org/10.1186/s13062-016-0134-5
- Gallardo R, Ramakers M, De Smet F, Claes F, Khodaparast L, Khodaparast L, Couceiro JR, Langenberg T, Siemons M, Nyström S, et al. De novo design of a biologically active amyloid. Science 2016; 354(6313):aah4949; PMID:27846578; http://dx.doi.org/10.1126/science.aah4949