
Abstract
Immunoglobulin G (IgG) has an unusually long serum half-life in comparison to proteins of a similar size. It is well-known that this phenomenon is due to IgG's ability to bind the neonatal Fc receptor (FcRn) in a pH-dependent manner. FcRn binding properties can vary among IgGs, resulting in altered in vivo half-lives, and therefore it would be beneficial to accurately predict the FcRn binding properties of therapeutic IgG monoclonal antibodies (mAbs). Here we describe the development of an in vitro model capable of predicting the in vivo half-life of human IgG. Using a high-throughput biolayer interferometry (BLI) platform, the human FcRn association rate at acidic pH and subsequent dissociation rate at physiological pH was determined for 5 human IgG1 mAbs. Comparing the combined FcRn association and dissociation rates to the Phase 1 clinical study half-lives of the mAbs resulted in a strong correlation. The correlation was also verified in vivo using mice transgenic for human FcRn. The model was used to characterize various factors that may influence FcRn-mAb binding, including mAb variable region sequence differences and constant region glycosylation patterns. Results indicated that the complementarity-determining regions of the heavy chain significantly influence the mAb's FcRn binding properties, while the absence of glycosylation does not alter mAb-FcRn binding. Development of this high-throughput FcRn binding model could potentially predict the half-life of therapeutic IgGs and aid in selection of lead candidates while also serving as a screening tool for the development of mAbs with desired pharmacokinetic properties.
Abbreviations
| mAb | = | monoclonal antibody |
| PK | = | pharmacokinetic |
| IgG | = | immunoglobulin G |
| FcRn | = | neonatal Fc receptor |
| β2m | = | β2-microglobulin |
| Fc | = | antibody constant region |
| Fab | = | antibody antigen binding fragment |
| hFcRn | = | human FcRn |
| BLI | = | biolayer interferometry |
| CDRs | = | complementary determining regions |
| VH | = | antibody heavy chain variable region |
| Vk | = | antibody light chain variable region |
| kon | = | association rate |
| koff | = | dissociation rate |
| KD | = | equilibrium affinity |
| CHO | = | Chinese Hamster Ovarian cells |
| ELISA | = | enzyme-linked immunosorbent assay |
| PNPP | = | para-nitrophenylphosphate |
Introduction
Since the first approval of products in the late 1980s and early 1990s, monoclonal antibodies (mAbs) have developed into the highest earning category of biological therapeutics.Citation1 While inflammatory disorders and cancer remain the main targets, mAb therapies are being considered for a variety of new indications and an increasing number of mAbs are receiving orphan drug status.Citation2-4 To meet the demand for mAbs to new targets and maintain competitiveness within the sector, novel discovery platforms and antibody engineering are required to develop the next generation of mAbs.Citation3,5-7 The pharmacokinetic (PK) properties of a mAb is widely regarded as a critical parameter to optimize for the successful production of more effective, safer and cheaper mAb therapeutics.Citation5,8-12
Fortunately, the last decade of research has enabled better understanding of biochemical and biophysical properties that can significantly influence mAb PK.Citation13 Aside from target-mediated clearance, which contributes to non-linear PK profiles and has limited opportunity for modulation,Citation14-16 the properties affecting linear and non-linear kinetics may include charge variation, nonspecific binding, size, delivery route, immunogenicity, post-translational modifications, hydrophobicity and proteolysis.Citation6,17-25 However, the ability for mAbs of the immunoglobulin G (IgG) isotype to bind the neonatal Fc receptor (FcRn) in a pH-dependent manner is the critical property associated with an IgG's linear kinetic profile and extends the half-life of a typical IgG1 to, on average, 21 d.Citation26-28 Functional FcRn, a heterodimeric complex of α-chain and β2-microglobulin (β2m),Citation29 was first discovered when the ability of IgG to transfer across the placental barrier from mother to fetus was observed.Citation30-32 Later, FcRn expression in the vascular endothelium and bone marrow-derived cells was attributed to the mechanism for IgG recycling and protection from lysosomal degradation.Citation33-36 The details of this mechanism have been widely investigated.Citation26,37-39 It is now well known that circulating IgGs are internalized by fluid phase pinocytosis and bind membrane-bound FcRn inside the acidified sorting endosomal compartments. Transport back to the cell surface and dissociation from FcRn at physiological pH rescues the IgG from catabolism and distributes it back to the circulation.
Elucidation of the crystal structure of FcRnCitation40-42 provided a means to investigate the IgG pH-dependent binding properties in detail and, as a result, a number of studies have defined the critical amino acid contacts.Citation43-47 More recently, mutations in these residues have exemplified the complex binding relationship of FcRn and IgG that modulates in vivo half-life, while highlighting the importance of both association at acidic pH (≈6.0) and dissociation at physiological pH (≈7.4).Citation11,18,23,37,38,48-55 Independent of the direct mAb-FcRn contact within the IgG constant region (Fc), additional studies have determined the antigen binding fragment (Fab) can have a significant effect on the mAb-FcRn interaction and in vivo half-life.Citation25,56,57 This phenomenon explains why circulating IgGs with identical Fc regions can vary 2-3 fold in serum half-life even when the same antigen is targeted.Citation58-60
Despite the well-characterized mAb-FcRn interaction and its effect on in vivo half-life, several hurdles that limit the usefulness of this information in experimental testing still remain. For example, several studies have attempted to measure the mAb-FcRn interaction in vitro and draw a correlation to in vivo half-life, but the results have been largely mixed.Citation24,25,37,50,52,55,56,61-64 This creates a dependency on the available transgenic FcRn mice for an accurate model of preclinical PK assessment,Citation22,64,65 which is lower throughput and more costly when compared to in vitro assays. Thus, a better assay is needed to support the fast-paced mAb discovery platforms currently in use. Here we report a novel high-throughput in vitro model capable of predicting the human half-life of naturally occurring, fully human IgG mAbs. This model performs similar to in vivo models with transgenic FcRn mice, and was used to predict features that may influence mAb half-life, including complementarity-determining regions (CDRs) and Fc region glycosylation.
Results
Production of human FcRn and pH-dependent binding to human IgG1
Human FcRn (hFcRn) was produced in Chinese hamster ovary (CHO) cells (α-chain with C-terminal His-tag and β2m with C-terminal myc-tag) and purified with IgG Sepharose to isolate hFcRn in its functional heterodimer form. Analysis by gel electrophoresis and western blot under reducing and denaturing conditions showed the recombinant FcRn was >90% pure with both α-chain and β2m expressed in equal proportions at the expected molecular weight (). In addition, western blots under native conditions showed that >85% of the purified recombinant protein was associated as an α-chain/β2m heterodimer at the expected molecular weight ().
Figure 1. Production and characterization of functional human FcRn heterodimer. SDS-PAGE and western blot under reducing and denaturing conditions (A) of purified recombinant hFcRn revealed pure material at the expected molecular weight (α-chain with his-tag = 32 kDa; β2m with myc-tag = 16 kDa). Western blot under native conditions (B) indicated the purified protein is in the expected α-chain/β2m heterodimer form at approximately 52 kDa. Interaction of the recombinant FcRn and a human IgG1 mAb (CDA1) is pH dependent (C). At acidic pH (6.0), CDA1 rapidly associated (kon) with FcRn, but was undetectable at physiological pH (7.5). Conversely, dissociation (kdis) of the mAb-FcRn complex was rapid at physiological pH, but slow at acidic pH.
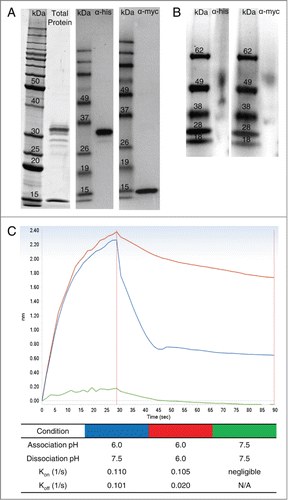
To verify the mAb-FcRn binding properties in a biolayer interferometry (BLI)-based assay format, an hFcRn-loaded surface was incubated with a human IgG1 antibody as the analyte at acidic pH (6.0) or physiological/slightly basic pH (7.5). Binding and dissociation rates were analyzed as described in Materials and Methods. Incubation at acidic pH resulted in rapid association (kon = 0.110 1/s), while physiological pH prevented any measurable binding (). Following association at acidic pH, the mAb-FcRn complex dissociated rapidly at pH 7.5 (koff = 0.101 1/s), while incubation at pH 6.0 resulted in a 5-fold slower off-rate ().
Correlation of human half-life to FcRn binding rate
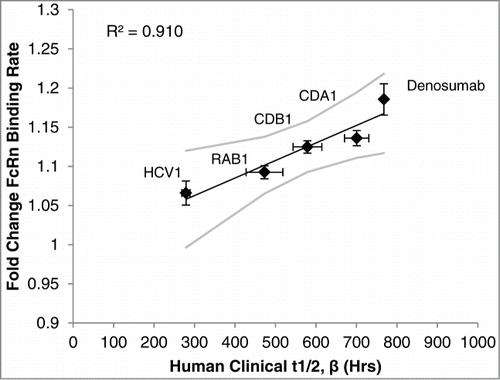
Both association and dissociation rates are important for FcRn-mediated recycling of IgG.Citation25 As such, we measured the association rate of the IgG-FcRn complex at acidic pH (6.0), followed by the dissociation rate at physiological pH (7.5) of 5 human IgG1s with a wide dynamic range of half-lives (∼12 to 32 days). The mAb association and dissociation rates were independently converted to fold change values relative to one another, and then the association and dissociation fold change values of a mAb were averaged to obtain the “fold change FcRn binding rate” for the given mAb. For example, CDA1 had an average association rate of 0.2087 ± 0.0036 1/s (± standard deviation), which was 1.0905 ± 0.0289 fold higher than the association rate of the lowest mAb assessed (RAB1), and an average dissociation rate of 0.0906 ± 0.0034 1/s, which was 1.1814 ± 0.0282 fold higher than the dissociation rate of the lowest mAb assessed (CDB1). Therefore, the average fold change FcRn binding rate for CDA1 was 1.1359 ± 0.0193. As seen in , plotting the experimentally derived fold change FcRn binding rate against the β phase half-life determined previously in Phase 1 clinical studies yielded a strong linear correlation (R2 = 0.910).
Figure 2. FcRn binding rate determined in vitro was compared to the human half-life previously determined in Phase 1 clinical studies. Using the BLI platform described in Materials and Methods, the fold change FcRn binding rate of 5 human IgG1 mAbs was independently determined and plotted against its known human half-life to establish a linear correlation. Values are the mean ± SEM. Line of best fit is shown in black with 95% confidence interval in gray. For in vitro experiments the average of at least 3 independent experiments is shown (N = 5 within each independent experiment). See Materials and Methods for details of clinical data.
Furthermore, the linear-curve fit model of the FcRn binding rate vs. human clinical half-life was exploited to back-calculate the predicted half-life of the mAbs in the in vitro model. The predicted half-life was within ∼10% of the clinical half-life for each mAb, and was within the error of the clinical study data ().
Table 1. Antibody half-lives predicted in vitro and clinically determined mAb half-lives
Predicting half-life of human mAbs
Given the predictive power of our model based on the 5 antibodies in , we assessed the ability of the predictive model to estimate half-life of antibodies in pre-clinical studies. The four lead mAbs of an ongoing project (mAbs A, B, C and D) were assessed using the in vitro model, as well as in vivo using hFcRn transgenic mice. The five standard mAbs (Denosumab, CDA1, CDB1, RAB1 and HCV1) were also assessed in vivo to verify the accuracy of the hFcRn mouse model and create a standard curve to calculate the in vivo predicted human half-life. In agreement with previous literature reports,Citation22,25 a strong positive linear correlation (R2=0.963) was detected between the observed half-life in hFcRn mice and the reported human half-life in clinical studies ().
Figure 3. IgG1 half-life in hFcRn transgenic mice to predict human half-life. The β phase half-life of the 5 standard mAbs in was assessed in hFcRn mice, which correlated to the actual human half-life determined in clinical studies (A). Linear regression analysis of the mAbs with known human half-life provided a line of best fit equation that was used to predict the human half-life of the pre-clinical experimental mAbs (denoted mAbs A-D) by applying the hFcRn mouse half-lives to the equation. The predicted human half-life using the hFcRn mouse in vivo data closely associated with the in vitro-predicted human half-life from the BLI-based assay (B). Values are the mean ± SEM. Line of best fit is shown in black with 95% confidence interval in gray. For mouse studies, 2–3 independent experiments were performed with N = 6 mice per experiment. In vitro results represent at least 2 independent experiments with N = 5 per experiment.
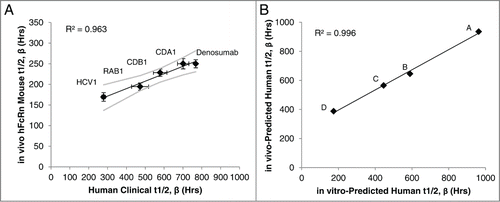
Based on the in vivo linear correlation between transgenic murine and human half-life, the line of best fit equation of the standard mAbs () and experimentally derived hFcRn mouse half-life was used to calculate the in vivo model prediction of human half-life for the 4 experimental mAbs. Similarly, the in vitro assay line of best fit equation from and experimentally derived fold change FcRn binding rate of the experimental mAbs was used to calculate the in vitro model predicted human half-life. The in vivo and in vitro model predictions closely associated with one another (R2 = 0.996; ), indicating both models predicted relative differences among the experimental mAbs similarly. Absolute values of the in vivo and in vitro predicted half-lives for the experimental mAbs plotted in are shown in , and also agree between the 2 models.
Table 2. Predicted human half-life of preclinical experimental mAbs using in vivo and in vitro models
Predicting half-life of modified antibodies
Primary amino acid sequence
To assess the ability of our in vitro predictive model to estimate the half-life of a given mAb following changes to the primary amino acid sequence, we decided to transplant the CDRs of one mAb (donor mAb) into the framework of another mAb (acceptor mAb) for the heavy (VH) or light chain (Vk). Several factors were considered for this experiment: 1) The donor and acceptor mAb had identical Fc region sequences; 2) The VH or Vk regions were of the same gene family, but contained several framework amino acid differences (13 for VH – 7 of which are similar amino acids; and 6 for Vk – 3 of which are similar amino acids); 3) The wild type donor and acceptor mAbs have significantly different half-lives in both Phase 1 clinical studies and the predictive model; and 4) The overall charge (specifically the pI) of the mAb was unchanged after exchanging the CDRs. Following these conditions, we could accurately predict the Fab region that influences the half-life of a mAb: the VH CDRs, VH framework mutations, Vk CDRs or Vk framework mutations.
Analysis using our in vitro predictive model indicated that inserting the VH CDRs of a mAb with a naturally longer half-life (CDA1) into the VH framework of a mAb with a significantly shorter half-life (HCV1) created a mutant mAb with a half-life predicted to be similar to the donor and significantly longer than the acceptor mAb (). The converse was also true, where insertion of VH CDRs of HCV1 into the framework of CDA1 created a mutant mAb with a predicted half-life similar to HCV1 and significantly shorter than CDA1 ().
Figure 4. Exchanging CDRs between mAbs to modulate FcRn-binding properties. Three wild type (WT) antibodies have distinct half-lives determined from clinical studies and were predicted using the in vitro BLI-based platform. Substitution of the 3 heavy chain (VH) complimentary determining regions (CDRs) significantly changed the predicted half-life of the mutant mAb to resemble the predicted half-life of the CDRs' parent mAb. Conversely, substitution of light chain (Vk) CDRs did not affect the predicted half-life. Substitution of both VH and Vk CDRs produced a mAb with a predicted half-life similar to the original half-life of the CDR parent mAb. Mean ± SD is reported of at least 2 independent experiments, with N = 5 for each experiment. Significant differences among mAb constructs are denoted by: *p < 0.05 vs. HCV-WT; @p < 0.05 vs. CDA-WT; #p < 0.05 vs. CDB-WT.
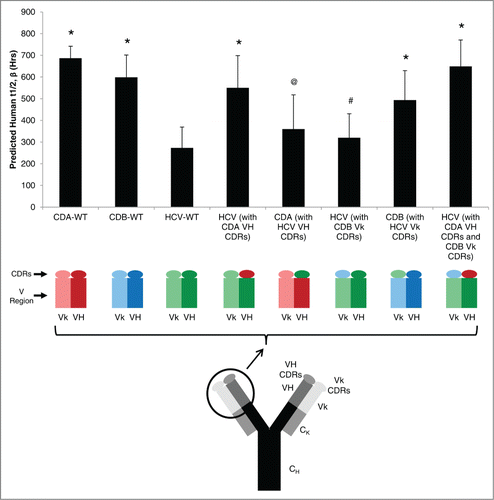
However, the influence of CDR sequence was not observed when the same approach was taken with Vk CDRs. In this case, inserting Vk CDRs of a mAb with a naturally longer half-life (CDB1) into the Vk framework of a mAb with a significantly lower half-life (HCV1) created a mutant mAb with a half-life similar to the acceptor mAb and significantly shorter than the donor mAb (). Again, the converse was true whereby the predicted half-life of the mutant mAb was significantly longer than the donor mAb following Vk substitution with CDRs from a mAb with a shorter half-life. Finally, creating a double mutant by inserting both VH and Vk CDRs from mAbs with longer half-lives into a mAb with a shorter half-life indicated the half-life of the double mutant adopted that of the donor mAbs (). Taken together, these experiments indicate that the VH CDRs represent the Fab region that significantly affects the half-life of a given mAb.
Post-translational modifications
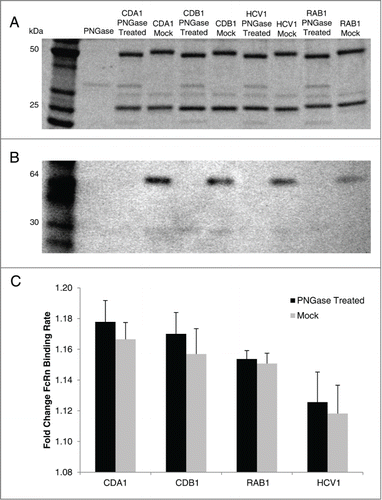
Modification of glycan structure is a common posttranslational event in vitro and in vivo. It has been reported that the absence of an Fc glycan in the germline CH2 domain does not alter the half-life of antibodies,Citation66-69 and thus we were interested in confirming this conclusion with our predictive model. Following PNGase treatment, 4 antibodies were confirmed to be aglycosylated by size shift and western blot analysis (). Comparison of hFcRn binding rates using the in vitro predictive model indicated no significant difference between glycosylated and aglycosylated mAb species (), confirming that this model is capable of portraying effects (or lack thereof) from post-translational modification.
Figure 5. FcRn binding properties of glycosylated or aglycosylated mAbs. PNGase treatment of 4 human IgG1 mAbs expressed in CHO cells produced aglycosylated versions that, when compared to mock treated mAb, was confirmed by size shift on SDS-PAGE (A) and lack of staining by Concanavalin A on western blot (B). Using the in vitro BLI-based assay to assess mAb-FcRn interactions, the glycosylated (mock) and aglycosylated (PNGase treated) version had similar binding rates in each case (C). Mean ±SD is reported for 3 independent experiments with N = 5 for each experiment.
Discussion
PK properties are often critical to the success of a therapeutic, and recent reports have suggested ∼15% of Phase 1 clinical studies fail due to inadequate PK or pharmacodynamics properties.Citation70 This is especially true for antibodies, where typically longer half-lives are desired for therapeutics to decrease dosing frequency and increase efficacy. Conversely, a short half-life may be desired for in vivo diagnostic mAbs where toxicity and off-target effects should be reduced. Here, we developed a novel in vitro, high-throughput screening method for predicting antibody half-life using a BLI platform.
Initially, high quality, fully functional human α-FcRn/β2m (hFcRn) was purified and characterized by its ability to bind human IgG1 in a pH-dependent manner. As expected, and consistent with literature reports,Citation28,55,57 a human monoclonal IgG1 associated quickly with hFcRn at a slightly acidic pH, while negligible binding was detected at physiological pH. Furthermore, dissociation of the mAb-hFcRn complex was slow at acidic pH and rapid at physiological pH. Similar to previous studies that conveyed the importance of high-quality hFcRn and assay format,Citation57 our results indicated the hFcRn we used is of high quality, and functions similar to its in vivo mechanism when positioned correctly as the ligand in a BLI platform. Neuber et al.Citation57 previously verified the accuracy of the BLI platform in comparison to surface plasmon resonance-based assays and highlighted the importance of further developing predictive models.
To establish a predictive model that considers both association and dissociation of the mAb-FcRn complex, a BLI kinetic model was used to calculate the association rate (kon) at acidic pH followed by the dissociation rate (koff) at physiological pH for 5 different antibodies with known half-lives ranging from ∼12 to ∼32 days; a generally acceptable dynamic range for therapeutic mAbs. A strong correlation between the FcRn binding rate and the human half-life determined from Phase 1 clinical studies was observed. Specifically, mAbs with a fast on or off rate had a longer half-life. Initial analysis indicated that both association and dissociation should be considered, as neither condition alone could positively correlate to the known human half-life (data not shown). These results may explain why some studies struggled to find a correlation between pH 6.0 equilibrium affinity and in vivo half-life.Citation24,37,50,52,56,61-64 Some predictive models have suggested mAb-FcRn equilibrium affinity (KD) at acidic pH is insufficient to accurately account for changes in half-life, and instead the rate of association should be considered.Citation49 Furthermore, other literature reports stress the importance of assessing dissociation characteristics at physiological pH to more accurately predict half-life.Citation25,37,55,63 It is likely that both the rates of association in the endosome and extracellular dissociation of the mAb-FcRn complex are critical for recycling. Our results support the use of on/off rates rather than equilibrium binding while considering the association and dissociation at relevant pH to accurately predict in vivo half-life.
To verify our model and extend its usefulness, we determined the predicted human half-life using the in vitro model for 4 lead human IgG1 mAbs still in preclinical studies, and compared the prediction to their in vivo half-life using hFcRn transgenic mice. This mouse model is widely accepted as an accurate surrogate for determining relative human half-lives of IgGs,Citation22,25 and our model confirmed the correlation between human and transgenic mouse half-life for the 5 mAbs tested with human clinical study data. Furthermore, comparing the estimated human half-life extrapolated from the in vivo study to the in vitro predicted half-life yielded a very strong positive correlation for the mAbs within a half-life range of ∼12–40 d Not only did the 4 antibodies have the same rank order in both assays, but the absolute predicted half-life varied by only about 3–20% between the 2 predictions for 3 of the 4 mAbs. Taken together, these results suggest the in vitro assay described here is capable of predicting the human half-life of mAbs similar to hFcRn transgenic mice.
A reliable, high-throughput assay that can predict the human half-life of a mAb after it has under gone modifications would be beneficial. To assess this possibility, and to address the question of what Fab region influences mAb half-life,Citation25,56,57 we decided to substitute CDR regions of one mAb with that of another. The in vitro assay was able to predict significantly different half-lives in select mutants and supported the hypothesis that heavy chain CDRs represent the region that influences mAb half-life as opposed to framework mutations or light chain CDRs. This expands similar investigations by Neuber et al.Citation57 who assessed the KD at acidic and physiological pH of a panel of mAbs to determine that the different germline immunoglobulin gene families did not influence FcRn binding. They further concluded that CDR1 and CDR2 regions also had no effect, and proposed that VH/VL framework mutations or the CDR3 sequence was responsible for modulation of the FcRn interaction. When combined with our results, it can be inferred that the CDR3 sequence is the cause of FcRn binding variation among different mAbs with the same constant region; however, additional experimentation followed by in vivo studies are needed for confirmation. Also, given the recent finding that local charge distribution can affect mAb PK,Citation71 it would be interesting to expand upon the case study presented here with a panel of mAbs containing various charge distributions in the CDR3 region.
In addition to primary amino acid sequence changes, post-translational modifications were also of interest. Early reports were conflicted regarding effects of glycosylation on antibody half-life,Citation18,72-75 but our results support the most recent reports suggesting glycosylation does not affect FcRn-mediated PK.Citation66-69 In our study, this was confirmed by removing all N-glycosylation (only the natural germline Fc glycosylation site exists in the antibodies tested) and the in vitro FcRn binding measurement was used to compare the aglycosylated mAb to the naturally occurring glycosylated form produced in CHO cells. This finding further suggests the in vitro model described here is capable of recapitulating data produced from more complicated, expensive and time-consuming in vivo studies that were previously required for reliable PK data. However, additional posttranslational modifications, including various glycan species, should be tested to confirm our model.
One limitation of the present study is the restriction to antibodies that bind soluble targets and are not affected by target-mediated clearance, which is expected to be a source of clearance for mAbs targeting the cell surface or other endogenous antigens.Citation16,22 Since the assay only measures mAb-FcRn interactions, factors influencing mAb clearance independent of FcRn cannot be predicted and animal models typically can address some of these factors. The influence of many biophysical properties, such as pI, are well established,Citation17 and standard features of the mAb can be assessed in conjunction with FcRn binding properties to gain a complete understanding of the mAb's PK properties. Indeed, others have suggested a combination of biophysical properties is responsible for a given mAb's PK profile.Citation19,57 We suggest the in vitro model described here can provide a critical piece of the puzzle, given FcRn interactions are a major parameter.
The quality of antibody preparations must be considered in addition to FcRn. For example, antibodies with a propensity to aggregate or degrade must be stored appropriately because destabilization of the mAb will significantly affect its FcRn binding properties. Although the mAbs are in defined buffers when FcRn binding is measured, optimal purification and formulation buffers should be used prior to analysis. It may be possible to use the FcRn binding assay to screen purification and formulation conditions given its sensitivity, and future studies will address this property.
Additional analysis should also refine the ability of the assay to predict the half-life of mAbs with Fc mutations, engineered Fc regions or Fc fusion proteins. For example, some mutations that enhance in vivo half-life have delayed release and higher affinity at physiological pH (data not shown),Citation12 which significantly affects the overall FcRn binding rate measured in this study. Regardless, select mutations have been shown to significantly enhance the half-life in humans.Citation76 Conversely, some mutations that reduce half-life, including H435A, can significantly alter the FcRn association rate,Citation77 making an overall FcRn binding rate measurement difficult. Modulating the assay parameters or method of calculating FcRn binding rate may allow these mutants to be accurately assessed using this model.
In summary, we developed a high throughput in vitro assay to predict the FcRn-mediated aspect of half-life for human IgG1 antibodies in humans. The assay is comparable to hFcRn transgenic mice in predictive power, yet it takes only 1 to 2 hours for full analysis of multiple mAbs, costs a fraction of the operating cost and requires limited mAb quantities (<0.5 mg for multiple experiments). Using the model, we found that the VH CDRs represented the Fab region that influences mAb half-life, and also confirmed recent studies claiming aglycosylated mAbs have similar half-lives compared to their glycosylated form. Further development of the model should allow additional properties of mAbs to be assessed with precision, including stability and the effects of Fc point mutants, among others.
Materials and Methods
Human FcRn production
Genes encoding the soluble ectodomain of human FcRn α-chain (amino acids 34–290; GenBank Accession #AAH0734.1) and full-length β2m (GenBank Accession #AAH64910.1) without the signal peptide were codon optimized for synthesis (Genewiz). A C-terminal His6 tag or a C-terminal Myc tag was cloned in frame with the α-chain or β2m proteins, respectively. Subsequently, a single vector was produced for the simultaneous expression of soluble human αFcRn and β2m in CHO cells, suspension cells adapted from CHOK1 (ATCC, CCL−61).
The full length plasmid containing α and β subunits was stably transfected in CHO cells via electroporation using the Gene Pulser Xcell system (BioRad). Soluble FcRn expression was assessed by anti-tag enzyme-linked immunosorbent assays (ELISA) and the 8 highest expressing CHO cell clones during early growth were pooled and scaled. The FcRn-containing media was collected, centrifuged and filtered using 0.2 µm filters. The heterodimeric, functional FcRn protein was purified from the media using IgG Sepharose 6 Fast Flow (GE Healthcare, 52–2083–00) on an Econo Column (BioRad, 737–2522). Based on the pH-dependent affinity of FcRn to IgG, columns were washed with PBS pH 6.0 and FcRn eluted with PBS pH 8.0. Fractions were concentrated with centrifugal concentrators (Pierce, Thermo Scientific, 89885A) and purified FcRn was stored at −80°C as single-use aliquots.
Characterization of purified hFcRn
FcRn protein purity was analyzed by SDS-Tris-glycine polyacrylamide gel electrophoresis (Life Technologies) under reducing conditions followed by SYPRO ruby staining (Life Technologies) using the manufacturers' recommended protocol. FcRn protein quality was also analyzed by Western blot under native conditions using Tris-Bis NativePage (Life Technologies) or reduced and denatured conditions using SDS-Tris-glycine polyacrylamide gels and transferred onto PVDF membranes (EMD Millipore). Using standard Western blot procedures, FcRn was probed with either a mouse anti-myc or a mouse anti-his antibody, detected by a secondary anti-mouse-HRP antibody (Jackson ImmunoResearch) and developed with Amersham ECL Western Blotting Detection Reagent (GE Healthcare, RPN2106). All gels and blots were imaged on a ChemiDoc MP (BioRad) and band intensity and molecular weight was estimated with Image Lab Software (BioRad).
Monoclonal antibodies
All mAbs used for in vitro and in vivo predictive models are fully human IgG1s that bind an exogenous or endogenous soluble target in vivo. The standard mAbs used to generate curve fit models for the in vitro and in vivo predictive model have reported human in vivo half-life data in healthy individuals. Of these 5 mAbs, 4 were produced by MassBiologics:Citation78-80 CDA1 (i.e. actoxumab; Merck), CDB1 (i.e., bezlotoxumab; Merck), RAB1 and HCV1. Denosumab was produced by Amgen.Citation81 All mAbs were produced under GMP conditions with a CHO cell manufacturing cell line. The four pre-clinical mAbs are also human IgG1s that bind an exogenous soluble target and were manufactured, purified and formulated by MassBiologics under GMP-like conditions using similar methods as the clinical mAbs.
BLI-based assessment of mAb-FcRn interactions
The Octet QK (PALL/ForteBio) was used for all FcRn binding in vitro assays at 30°C in 96-well solid black plates (Greiner Bio-One, 655900). Initially, FcRn was immobilized to nickel-nitrilotriacetic acid -coated biosensors (ForteBio, 18–0029) for 180 seconds at an optimized concentration. After a baseline step, the mAb-FcRn binding rate was determined when the biosensor with immobilized FcRn was exposed to the mAb sample in PBS adjusted to pH 6.0 with HCl for 30 seconds. Following association, the mAb-FcRn complex was exposed to PBS pH 7.5 and the rate of mAb dissociation from FcRn was measured. Prior to analysis, all antibodies were dialyzed into PBS pH 6.0, diluted to 100 µg/ml in PBS pH 6.0 and used at a 200 µl volume. Each assay is performed on a specific mAb in quintuplicate. Data analysis was performed using software version 7.0 (PALL, ForteBio). In a step specific manner, the given Kon and Koff rates were determined using the first 20 and 30 seconds of the association and dissociation steps, respectively. Savitzky-Golay filtering was applied to increase signal-to-noise ratio and rates were calculated with R equilibrium steady-state fitting. Rates were reported in 1/s and the mean Kon and Koff values of each mAb within an experiment were individually normalized to fold-change values among all tested mAbs. The fold-change FcRn binding rate was determined by averaging the mean fold-change Kon and mean fold-change Koff values of a mAb. The mAb FcRn binding rates and the accompanying known human half-lives from human Phase 1 clinical study data were used to generate a linear regression best-fit correlation.
hFcRn mice in vivo half life prediction
Human FcRn transgenic mice used in this study were hemizygous B6.Cg-Fcgrttm1Dcr Tg(FCGRT)32Dcr/DcrJ from The Jackson Laboratory (Stock #014565). These animals lack the endogenous mouse FcRn α gene and express the human FcRn α gene on one allele under control of the native FcRn promoter. The animals have the native mouse β2m gene. The specific strain was selected based on a previous study examining the different strains.Citation22 Female mice of 3 different age groups were used in this study: 8, 14 and 20 weeks old.
For PK studies, mice received a single intraperitoneal injection containing a mixture of IgGs such that each mAb was administered at 1 mg/kg. Each mixture was composed of the 5 mAbs with known human half-life (denosumab, CDA1, CDB1, HCV1 and RAB1) and 4 or 5 different pre-clinical mAbs. All mAbs within a mixture could be individually identified by specific antigen binding. Serial submandibular bleeds were collected in Microtainer Serum Separator Tubes (BD, 365956) at 2 hours, 1 day, 7 days, 14 days, 21 days, 29 d and 41 d after injection. Animals were separated into subgroups to stagger early time point bleeds. Blood samples were centrifuged at 3500 rpm for 10 minutes and the serum fraction was collected and stored at −80°C until use.
The concentration of each human IgG in mouse serum samples at the various time points was determined by ELISA. Briefly, 96-well High-binding ELISA plates (Costar, 07–200–39) were coated with an optimized concentration of the mAb-specific antigen in PBS and blocked with 1% Bovine Albumin Fraction V (MP Biomedicals, 810034) in PBS + 0.05% Tween20 (Sigma-Aldrich, P1379). Mouse sera samples and standards (mAb mix used for injections or single mAb alone) were serially diluted in blocking buffer and incubated on antigen-coated plates for 1 hour. Bound hIgG was detected with Alkaline Phosphatase-conjugated AffiniPure Goat Anti-Human IgG (Jackson ImmunoResearch, 109–055–098) after developing with para-nitrophenylphosphate (PNPP) (Thermo, 34047). Optical density (OD) was measured at 405nm on a Precision Microplate Reader (Molecular Devices) and mean mAb concentration of test samples were determined from the standard curve using a 4-parameter nonlinear regression model in Softmax Pro software (Molecular Devices). Extrapolated concentrations were multiplied by sera dilution factors and the mean value was used to determine final IgG concentrations.
To identify mice that mounted an anti-hIgG immune response during the experiment, plates were coated with the mAb mix used for injection diluted in PBS to 1µg/mL per mAb. Plates were blocked and sera samples from day 21 to 41 time points were serially diluted and added to the plate. Bound murine IgG was detected with Alkaline Phosphatase-conjugated AffiniPure Goat Anti-Mouse IgG (Jackson ImmunoResearch, 115–055–003) after developing with PNPP (Thermo, 34047). OD was measured at 405nm on Precision Microplate Reader (Molecular Devices) and mice with ODs 5-fold background levels were considered to have a significant anti-hIgG response and were removed from data analysis.
Half-life calculations were determined using Phoenix WinNonlin 6.3 (Certara) noncompartmental analysis. Consistent time points representing the β-phase half-life (7–41 d post injection) were analyzed using the linear trapezoidal linear interpolation calculation method. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee of the University of Massachusetts Medical School (Protocol Number: A-2412).
CDR-exchanged mAbs
Mutant mAb gene sequences were constructed using the IMGT database to identify CDR region nucleotides within the heavy and light chain variable regions. Differences between mAbs in the framework region primary amino acid sequence were identified using the alignment tool in Vector NTI (Life Technologies) with the default Similarity Table settings. Mutant sequences were constructed such that the variable region contained the framework of one mAb (denoted the “acceptor” mAb) and the CDR1, 2 and 3 of a different mAb (denoted the “donor” mAb). The modified genes were synthesized (Genewiz) and heavy and light chain genes were transiently co-transfected in 293T Human Endothelial Kidney Cells (ATCC, CRL-11268) with 25 kDa linear polyethylenimine (Polysciences). The media was collected 6 d after transfection and centrifuged to remove cell debris. The IgG-containing media was filtered using 0.2 µm filters (Thermo Scientific, Nalgene), purified by standard Protein A sepharose affinity methods and eluted with 100 mM glycine pH 2.8. Purified mAbs were dialyzed into PBS pH 6.0 and stored at 4°C until use.
Aglycosylated mAbs
Four mAbs produced by MassBiologics (CDA1, CDB1, HCV1, and RAB1) were treated with PNGase F (New England BioLabs) to remove N-linked glycosylation. For each antibody, 100µg was incubated with 6µl of PNGase F for 3 hours at 37°C. A mock control reaction was performed with 6µl of PNGase buffer instead of enzyme. The product was analyzed by SDS-Tris-glycine PAGE (Life Technologies) under reducing/denaturing conditions and SYPRO ruby stained (Life Technologies) using the manufacturers' protocol. The absence of N-linked glycan was also analyzed by Western blot after transfer to PVDF membrane (EMD Millipore). The blot was probed with a 1µg/ml solution of HRP conjugated Concanavilin-A (Sigma-Aldrich) and developed with Amersham ECL Western Blotting Detection Reagent (GE Healthcare, RPN2106).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Leila Sevigny, Brian Booth, Gregory Babcock and Teresa Broering for project initiation and contribution of ideas, Chloe Emery for her assistance with hFcRn mice and the MassBiologics Process Development department for production, purification and formulation of antibodies.
Funding
Research was funded in part by the Defense Advanced Research Projects Agency (DARPA-BAA-13–03).
References
- Aggarwal S. What's fueling the biotech engine–2010 to 2011. Nat Biotechnol 2011; 29:1083-9; PMID:22158359; http://dx.doi.org/10.1038/nbt.2060
- Reichert JM. Antibodies to watch in 2014. MAbs 2014; 6:5-14; PMID:24284914; http://dx.doi.org/10.4161/mabs.27333
- Beck A, Wurch T, Bailly C, Corvaia N. Strategies and challenges for the next generation of therapeutic antibodies. Nat Rev Immunol 2010; 10:345-52; PMID:20414207; http://dx.doi.org/10.1038/nri2747
- Murad JP, Lin OA, Espinosa EV, Khasawneh FT. Current and experimental antibody-based therapeutics: insights, breakthroughs, setbacks and future directions. Curr Mol Med 2013; 13:165-78; PMID:22834842; http://dx.doi.org/10.2174/156652413804486322
- Chan AC, Carter PJ. Therapeutic antibodies for autoimmunity and inflammation. Nat Rev Immunol 2010; 10:301-16; PMID:20414204; http://dx.doi.org/10.1038/nri2761
- Igawa T, Tsunoda H, Kuramochi T, Sampei Z, Ishii S, Hattori K. Engineering the variable region of therapeutic IgG antibodies. MAbs 2011; 3:243-52; PMID:21406966; http://dx.doi.org/10.4161/mabs.3.3.15234
- Carter PJ. Introduction to current and future protein therapeutics: a protein engineering perspective. Exp Cell Res 2011; 317:1261-9; PMID:21371474; http://dx.doi.org/10.1016/j.yexcr.2011.02.013
- Leavy O. Therapeutic antibodies: past, present and future. Nat Rev Immunol 2010; 10:297; PMID:20422787; http://dx.doi.org/10.1038/nri2763
- Carter PJ. Potent antibody therapeutics by design. Nat Rev Immunol 2006; 6:343-57; PMID:16622479; http://dx.doi.org/10.1038/nri1837
- Presta LG. Molecular engineering and design of therapeutic antibodies. Curr Opin Immunol 2008; 20:460-70; PMID:18656541; http://dx.doi.org/10.1016/j.coi.2008.06.012
- Zalevsky J, Chamberlain AK, Horton HM, Karki S, Leung IW, Sproule TJ, Lazar GA, Roopenian DC, Desjarlais JR. Enhanced antibody half-life improves in vivo activity. Nat Biotechnol 2010; 28:157-9; PMID:20081867; http://dx.doi.org/10.1038/nbt.1601
- Ko SY, Pegu A, Rudicell RS, Yang ZY, Joyce MG, Chen X, Wang K, Bao S, Kraemer TD, Rath T, et al. Enhanced neonatal Fc receptor function improves protection against primate SHIV infection. Nature 2014; 514:642-5; PMID:25119033; http://dx.doi.org/10.1038/nature13612
- Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci 2004; 93:2645-68; PMID:15389672; http://dx.doi.org/10.1002/jps.20178
- Igawa T, Mimoto F, Hattori K. pH-dependent antigen-binding antibodies as a novel therapeutic modality. Biochim Biophys Acta 2014; 1844:1943-50; PMID:25125373; http://dx.doi.org/10.1016/j.bbapap.2014.08.003
- Igawa T, Ishii S, Tachibana T, Maeda A, Higuchi Y, Shimaoka S, Moriyama C, Watanabe T, Takubo R, Doi Y, et al. Antibody recycling by engineered pH-dependent antigen binding improves the duration of antigen neutralization. Nat Biotechnol 2010; 28:1203-7; PMID:20953198; http://dx.doi.org/10.1038/nbt.1691
- Tabrizi M, Bornstein GG, Suria H. Biodistribution mechanisms of therapeutic monoclonal antibodies in health and disease. AAPS J 2010; 12:33-43; PMID:19924542; http://dx.doi.org/10.1208/s12248-009-9157-5
- Igawa T, Tsunoda H, Tachibana T, Maeda A, Mimoto F, Moriyama C, Nanami M, Sekimori Y, Nabuchi Y, Aso Y, et al. Reduced elimination of IgG antibodies by engineering the variable region. Protein Eng Des Sel 2010; 23:385-92; PMID:20159773; http://dx.doi.org/10.1093/protein/gzq009
- Bumbaca D, Boswell CA, Fielder PJ, Khawli LA. Physiochemical and biochemical factors influencing the pharmacokinetics of antibody therapeutics. AAPS J 2012; 14:554-8; PMID:22610647; http://dx.doi.org/10.1208/s12248-012-9369-y
- Datta-Mannan A, Chow CK, Dickinson C, Driver D, Lu J, Witcher DR, Wroblewski VJ. FcRn affinity-pharmacokinetic relationship of five human IgG4 antibodies engineered for improved in vitro FcRn binding properties in cynomolgus monkeys. Drug Metab Dispos 2012; 40:1545-55; PMID:22584253; http://dx.doi.org/10.1124/dmd.112.045864
- Keizer RJ, Huitema AD, Schellens JH, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet 2010; 49:493-507; PMID:20608753; http://dx.doi.org/10.2165/11531280-000000000-00000
- Khawli LA, Goswami S, Hutchinson R, Kwong ZW, Yang J, Wang X, Yao Z, Sreedhara A, Cano T, Tesar D, et al. Charge variants in IgG1: Isolation, characterization, in vitro binding properties and pharmacokinetics in rats. MAbs 2010; 2:613-24; PMID:20818176; http://dx.doi.org/10.4161/mabs.2.6.13333
- Tam SH, McCarthy SG, Brosnan K, Goldberg KM, Scallon BJ. Correlations between pharmacokinetics of IgG antibodies in primates vs. FcRn-transgenic mice reveal a rodent model with predictive capabilities. MAbs 2013; 5:397-405; PMID:23549129; http://dx.doi.org/10.4161/mabs.23836
- Hinton PR, Xiong JM, Johlfs MG, Tang MT, Keller S, Tsurushita N. An engineered human IgG1 antibody with longer serum half-life. J Immunol 2006; 176:346-56; PMID:16365427; http://dx.doi.org/10.4049/jimmunol.176.1.346
- Datta-Mannan A, Witcher DR, Tang Y, Watkins J, Wroblewski VJ. Monoclonal antibody clearance. Impact of modulating the interaction of IgG with the neonatal Fc receptor. J Biol Chem 2007; 282:1709-17; PMID:17135257; http://dx.doi.org/10.1074/jbc.M607161200
- Wang W, Lu P, Fang Y, Hamuro L, Pittman T, Carr B, Hochman J, Prueksaritanont T. Monoclonal antibodies with identical Fc sequences can bind to FcRn differentially with pharmacokinetic consequences. Drug Metab Dispos 2011; 39:1469-77; PMID:21610128; http://dx.doi.org/10.1124/dmd.111.039453
- Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 2007; 7:715-25; PMID:17703228; http://dx.doi.org/10.1038/nri2155
- Kacskovics I, Cervenak J, Erdei A, Goldsby RA, Butler JE. Recent advances using FcRn overexpression in transgenic animals to overcome impediments of standard antibody technologies to improve the generation of specific antibodies. MAbs 2011; 3:431-9; PMID:22048692; http://dx.doi.org/10.4161/mabs.3.5.17023
- Rodewald R. pH-dependent binding of immunoglobulins to intestinal cells of the neonatal rat. J Cell Biol 1976; 71:666-9; PMID:11223; http://dx.doi.org/10.1083/jcb.71.2.666
- Praetor A, Hunziker W. β(2)-Microglobulin is important for cell surface expression and pH-dependent IgG binding of human FcRn. J Cell Sci 2002; 115:2389-97; PMID:12006623
- Morphis LG, Gitlin D. Maturation of the maternofoetal transport system for human gamma-globulin in the mouse. Nature 1970; 228:573; PMID:5528694; http://dx.doi.org/10.1038/228573a0
- Brambell FW. The transmission of immunity from mother to young and the catabolism of immunoglobulins. Lancet 1966; 2:1087-93; PMID:4162525; http://dx.doi.org/10.1016/S0140-6736(66)92190-8
- Ghetie V, Ward ES. Multiple roles for the major histocompatibility complex class I- related receptor FcRn. Annu Rev Immunol 2000; 18:739-66; PMID:10837074; http://dx.doi.org/10.1146/annurev.immunol.18.1.739
- Ward ES, Zhou J, Ghetie V, Ober RJ. Evidence to support the cellular mechanism involved in serum IgG homeostasis in humans. Int Immunol 2003; 15:187-95; PMID:12578848; http://dx.doi.org/10.1093/intimm/dxg018
- Borvak J, Richardson J, Medesan C, Antohe F, Radu C, Simionescu M, Ghetie V, Ward ES. Functional expression of the MHC class I-related receptor, FcRn, in endothelial cells of mice. Int Immunol 1998; 10:1289-98; PMID:9786428; http://dx.doi.org/10.1093/intimm/10.9.1289
- Antohe F, Radulescu L, Gafencu A, Ghetie V, Simionescu M. Expression of functionally active FcRn and the differentiated bidirectional transport of IgG in human placental endothelial cells. Hum Immunol 2001; 62:93-105; PMID:11182218; http://dx.doi.org/10.1016/S0198-8859(00)00244-5
- Akilesh S, Christianson GJ, Roopenian DC, Shaw AS. Neonatal FcR expression in bone marrow-derived cells functions to protect serum IgG from catabolism. J Immunol 2007; 179:4580-8; PMID:17878355; http://dx.doi.org/10.4049/jimmunol.179.7.4580
- Dall'Acqua WF, Woods RM, Ward ES, Palaszynski SR, Patel NK, Brewah YA, Wu H, Kiener PA, Langermann S. Increasing the affinity of a human IgG1 for the neonatal Fc receptor: biological consequences. J Immunol 2002; 169:5171-80; PMID:12391234; http://dx.doi.org/10.4049/jimmunol.169.9.5171
- Vaccaro C, Zhou J, Ober RJ, Ward ES. Engineering the Fc region of immunoglobulin G to modulate in vivo antibody levels. Nat Biotechnol 2005; 23:1283-8; PMID:16186811; http://dx.doi.org/10.1038/nbt1143
- Kuo TT, Aveson VG. Neonatal Fc receptor and IgG-based therapeutics. MAbs 2011; 3:422-30; PMID:22048693; http://dx.doi.org/10.4161/mabs.3.5.16983
- Burmeister WP, Gastinel LN, Simister NE, Blum ML, Bjorkman PJ. Crystal structure at 2.2 A resolution of the MHC-related neonatal Fc receptor. Nature 1994; 372:336-43; PMID:7969491; http://dx.doi.org/10.1038/372336a0
- Burmeister WP, Huber AH, Bjorkman PJ. Crystal structure of the complex of rat neonatal Fc receptor with Fc. Nature 1994; 372:379-83; PMID:7969498; http://dx.doi.org/10.1038/372379a0
- West AP Jr., Bjorkman PJ. Crystal structure and immunoglobulin G binding properties of the human major histocompatibility complex-related Fc receptor(,). Biochemistry 2000; 39:9698-708; PMID:10933786; http://dx.doi.org/10.1021/bi000749m
- Raghavan M, Chen MY, Gastinel LN, Bjorkman PJ. Investigation of the interaction between the class I MHC-related Fc receptor and its immunoglobulin G ligand. Immunity 1994; 1:303-15; PMID:7889418; http://dx.doi.org/10.1016/1074-7613(94)90082-5
- Kim JK, Firan M, Radu CG, Kim CH, Ghetie V, Ward ES. Mapping the site on human IgG for binding of the MHC class I-related receptor, FcRn. Eur J Immunol 1999; 29:2819-25; PMID:10508256; http://dx.doi.org/10.1002/(SICI)1521-4141(199909)29:09%3c2819::AID-IMMU2819%3e3.0.CO;2-6
- Medesan C, Matesoi D, Radu C, Ghetie V, Ward ES. Delineation of the amino acid residues involved in transcytosis and catabolism of mouse IgG1. J Immunol 1997; 158:2211-7; PMID:9036967
- Oganesyan V, Damschroder MM, Cook KE, Li Q, Gao C, Wu H, Dall'Acqua WF. Structural insights into neonatal Fc receptor-based recycling mechanisms. J Biol Chem 2014; 289:7812-24; PMID:24469444; http://dx.doi.org/10.1074/jbc.M113.537563
- Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, Xie D, Lai J, Stadlen A, Li B, et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem 2001; 276:6591-604; PMID:11096108; http://dx.doi.org/10.1074/jbc.M009483200
- Ghetie V, Popov S, Borvak J, Radu C, Matesoi D, Medesan C, Ober RJ, Ward ES. Increasing the serum persistence of an IgG fragment by random mutagenesis. Nat Biotechnol 1997; 15:637-40; PMID:9219265; http://dx.doi.org/10.1038/nbt0797-637
- Chen Y, Balthasar JP. Evaluation of a catenary PBPK model for predicting the in vivo disposition of mAbs engineered for high-affinity binding to FcRn. AAPS J 2012; 14:850-9; PMID:22956476; http://dx.doi.org/10.1208/s12248-012-9395-9
- Deng R, Loyet KM, Lien S, Iyer S, DeForge LE, Theil FP, Lowman HB, Fielder PJ, Prabhu S. Pharmacokinetics of humanized monoclonal anti-tumor necrosis factor-{α} antibody and its neonatal Fc receptor variants in mice and cynomolgus monkeys. Drug Metab Dispos 2010; 38:600-5; PMID:20071453; http://dx.doi.org/10.1124/dmd.109.031310
- Dall'Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J Biol Chem 2006; 281:23514-24; PMID:16793771; http://dx.doi.org/10.1074/jbc.M604292200
- Datta-Mannan A, Witcher DR, Tang Y, Watkins J, Jiang W, Wroblewski VJ. Humanized IgG1 variants with differential binding properties to the neonatal Fc receptor: relationship to pharmacokinetics in mice and primates. Drug Metab Dispos 2007; 35:86-94; PMID:17050651; http://dx.doi.org/10.1124/dmd.106.011734
- Hinton PR, Johlfs MG, Xiong JM, Hanestad K, Ong KC, Bullock C, Keller S, Tang MT, Tso JY, Vasquez M, et al. Engineered human IgG antibodies with longer serum half-lives in primates. J Biol Chem 2004; 279:6213-6; PMID:14699147; http://dx.doi.org/10.1074/jbc.C300470200
- Monnet C, Jorieux S, Souyris N, Zaki O, Jacquet A, Fournier N, Crozet F, de Romeuf C, Bouayadi K, Urbain R, et al. Combined glyco- and protein-Fc engineering simultaneously enhance cytotoxicity and half-life of a therapeutic antibody. MAbs 2014; 6:422-36; PMID:24492301; http://dx.doi.org/10.4161/mabs.27854
- Borrok MJ, Wu Y, Beyaz N, Yu XQ, Oganesyan V, Dall'Acqua WF, Tsui P. pH-dependent Binding Engineering Reveals an FcRn Affinity Threshold That Governs IgG Recycling. J Biol Chem 2014; 290:4282-90; PMID:25538249; http://dx.doi.org/10.1074/jbc.M114.603712
- Suzuki T, Ishii-Watabe A, Tada M, Kobayashi T, Kanayasu-Toyoda T, Kawanishi T, Yamaguchi T. Importance of neonatal FcR in regulating the serum half-life of therapeutic proteins containing the Fc domain of human IgG1: a comparative study of the affinity of monoclonal antibodies and Fc-fusion proteins to human neonatal FcR. J Immunol 2010; 184:1968-76; PMID:20083659; http://dx.doi.org/10.4049/jimmunol.0903296
- Neuber T, Frese K, Jaehrling J, Jager S, Daubert D, Felderer K, Linnemann M, Hohne A, Kaden S, Kolln J, et al. Characterization and screening of IgG binding to the neonatal Fc receptor. MAbs 2014; 6:928-42; PMID:24802048; http://dx.doi.org/10.4161/mabs.28744
- Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther 2008; 84:548-58; PMID:18784655; http://dx.doi.org/10.1038/clpt.2008.170
- Zhu Y, Hu C, Lu M, Liao S, Marini JC, Yohrling J, Yeilding N, Davis HM, Zhou H. Population pharmacokinetic modeling of ustekinumab, a human monoclonal antibody targeting IL-12/23p40, in patients with moderate to severe plaque psoriasis. J Clin Pharmacol 2009; 49:162-75; PMID:19179295; http://dx.doi.org/10.1177/0091270008329556
- Gandhi M, Alwawi E, Gordon KB. Anti-p40 antibodies ustekinumab and briakinumab: blockade of interleukin-12 and interleukin-23 in the treatment of psoriasis. Semin Cutan Med Surg 2010; 29:48-52; PMID:20430307; http://dx.doi.org/10.1016/j.sder.2010.02.001
- Yeung YA, Wu X, Reyes AE 2nd, Vernes JM, Lien S, Lowe J, Maia M, Forrest WF, Meng YG, Damico LA, et al. A therapeutic anti-VEGF antibody with increased potency independent of pharmacokinetic half-life. Cancer Res 2010; 70:3269-77; PMID:20354184; http://dx.doi.org/10.1158/0008-5472.CAN-09-4580
- Gurbaxani B, Dela Cruz LL, Chintalacharuvu K, Morrison SL. Analysis of a family of antibodies with different half-lives in mice fails to find a correlation between affinity for FcRn and serum half-life. Mol Immunol 2006; 43:1462-73; PMID:16139891; http://dx.doi.org/10.1016/j.molimm.2005.07.032
- Yeung YA, Leabman MK, Marvin JS, Qiu J, Adams CW, Lien S, Starovasnik MA, Lowman HB. Engineering human IgG1 affinity to human neonatal Fc receptor: impact of affinity improvement on pharmacokinetics in primates. J Immunol 2009; 182:7663-71; PMID:19494290; http://dx.doi.org/10.4049/jimmunol.0804182
- Vaccaro C, Bawdon R, Wanjie S, Ober RJ, Ward ES. Divergent activities of an engineered antibody in murine and human systems have implications for therapeutic antibodies. Proc Natl Acad Sci U S A 2006; 103:18709-14; PMID:17116867; http://dx.doi.org/10.1073/pnas.0606304103
- Petkova SB, Akilesh S, Sproule TJ, Christianson GJ, Al Khabbaz H, Brown AC, Presta LG, Meng YG, Roopenian DC. Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: potential application in humorally mediated autoimmune disease. Int Immunol 2006; 18:1759-69; PMID:17077181; http://dx.doi.org/10.1093/intimm/dxl110
- Hristodorov D, Fischer R, Joerissen H, Muller-Tiemann B, Apeler H, Linden L. Generation and comparative characterization of glycosylated and aglycosylated human IgG1 antibodies. Mol Biotechnol 2013; 53:326-35; PMID:22427250; http://dx.doi.org/10.1007/s12033-012-9531-x
- Liu L, Stadheim A, Hamuro L, Pittman T, Wang W, Zha D, Hochman J, Prueksaritanont T. Pharmacokinetics of IgG1 monoclonal antibodies produced in humanized Pichia pastoris with specific glycoforms: a comparative study with CHO produced materials. Biologicals 2011; 39:205-10; PMID:21723741; http://dx.doi.org/10.1016/j.biologicals.2011.06.002
- Jung ST, Kang TH, Kelton W, Georgiou G. Bypassing glycosylation: engineering aglycosylated full-length IgG antibodies for human therapy. Curr Opin Biotechnol 2011; 22:858-67; PMID:21420850; http://dx.doi.org/10.1016/j.copbio.2011.03.002
- Millward TA, Heitzmann M, Bill K, Langle U, Schumacher P, Forrer K. Effect of constant and variable domain glycosylation on pharmacokinetics of therapeutic antibodies in mice. Biologicals 2008; 36:41-7; PMID:17890101; http://dx.doi.org/10.1016/j.biologicals.2007.05.003
- Cook D, Brown D, Alexander R, March R, Morgan P, Satterthwaite G, Pangalos MN. Lessons learned from the fate of AstraZeneca's drug pipeline: a five-dimensional framework. Nat Rev Drug Discov 2014; 13:419-31; PMID:24833294; http://dx.doi.org/10.1038/nrd4309
- Datta-Mannan A, Thangaraju A, Leung D, Tang Y, Witcher DR, Lu J, Wroblewski VJ. Balancing charge in the complementarity-determining regions of humanized mAbs without affecting pI reduces non-specific binding and improves the pharmacokinetics. mAbs 2015; 7:483-93; PMID:25695748
- Newkirk MM, Novick J, Stevenson MM, Fournier MJ, Apostolakos P. Differential clearance of glycoforms of IgG in normal and autoimmune-prone mice. Clin Exp Immunol 1996; 106:259-64; PMID:8918571; http://dx.doi.org/10.1046/j.1365-2249.1996.d01-847.x
- Wright A, Morrison SL. Effect of altered CH2-associated carbohydrate structure on the functional properties and in vivo fate of chimeric mouse-human immunoglobulin G1. J Exp Med 1994; 180:1087-96; PMID:8064227; http://dx.doi.org/10.1084/jem.180.3.1087
- Wright A, Sato Y, Okada T, Chang K, Endo T, Morrison S. In vivo trafficking and catabolism of IgG1 antibodies with Fc associated carbohydrates of differing structure. Glycobiology 2000; 10:1347-55; PMID:11159927; http://dx.doi.org/10.1093/glycob/10.12.1347
- Marian M, Harris R. The impact of Fc glycans on the clearance of Xolair. WCBP-congress 2002
- Robbie GJ, Criste R, Dall'acqua WF, Jensen K, Patel NK, Losonsky GA, Griffin MP. A novel investigational Fc-modified humanized monoclonal antibody, motavizumab-YTE, has an extended half-life in healthy adults. Antimicrob Agents Chemother 2013; 57:6147-53; PMID:24080653; http://dx.doi.org/10.1128/AAC.01285-13
- Firan M, Bawdon R, Radu C, Ober RJ, Eaken D, Antohe F, Ghetie V, Ward ES. The MHC class I-related receptor, FcRn, plays an essential role in the maternofetal transfer of gamma-globulin in humans. Int Immunol 2001; 13:993-1002; PMID:11470769; http://dx.doi.org/10.1093/intimm/13.8.993
- Chung RT, Gordon FD, Curry MP, Schiano TD, Emre S, Corey K, Markmann JF, Hertl M, Pomposelli JJ, Pomfret EA, et al. Human monoclonal antibody MBL-HCV1 delays HCV viral rebound following liver transplantation: a randomized controlled study. Am J Transplant 2013; 13:1047-54; PMID:23356386; http://dx.doi.org/10.1111/ajt.12083
- Lowy I, Molrine DC, Leav BA, Blair BM, Baxter R, Gerding DN, Nichol G, Thomas WD Jr., Leney M, Sloan S, et al. Treatment with monoclonal antibodies against Clostridium difficile toxins. N Engl J Med 2010; 362:197-205; PMID:20089970; http://dx.doi.org/10.1056/NEJMoa0907635
- Sloan SE, Hanlon C, Weldon W, Niezgoda M, Blanton J, Self J, Rowley KJ, Mandell RB, Babcock GJ, Thomas WD Jr., et al. Identification and characterization of a human monoclonal antibody that potently neutralizes a broad panel of rabies virus isolates. Vaccine 2007; 25:2800-10; PMID:17240489; http://dx.doi.org/10.1016/j.vaccine.2006.12.031
- Anastasilakis AD, Toulis KA, Polyzos SA, Anastasilakis CD, Makras P. Long-term treatment of osteoporosis: safety and efficacy appraisal of denosumab. Ther Clin Risk Manag 2012; 8:295-306; PMID:22767993; http://dx.doi.org/10.2147/TCRM.S24239