
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Bi-specific antibodies (BsAbs), which can simultaneously block 2 tumor targets, have emerged as promising therapeutic alternatives to combinations of individual monoclonal antibodies. Here, we describe the engineering and development of a novel, human bi-functional antibody-receptor domain fusion molecule with ligand capture (bi-AbCap) through the fusion of the domain 2 of human vascular endothelial growth factor receptor 1 (VEGFR1) to an antibody directed against insulin-like growth factor – type I receptor (IGF-IR). The bi-AbCap possesses excellent stability and developability, and is the result of minimal engineering. Beyond potent neutralizing activities against IGF-IR and VEGF, the bi-AbCap is capable of cross-linking VEGF to IGF-IR, leading to co-internalization and degradation of both targets by tumor cells. In multiple mouse xenograft tumor models, the bi-AbCap improves anti-tumor activity over individual monotherapies. More importantly, it exhibits superior inhibition of tumor growth, compared with the combination of anti-IGF-IR and anti-VEGF therapies, via powerful blockade of both direct tumor cell growth and tumor angiogenesis. The unique “capture-for-degradation” mechanism of the bi-AbCap is informative for the design of next-generation bi-functional anti-cancer therapies directed against independent signaling pathways. The bi-AbCap design represents an alternative approach to the creation of dual-targeting antibody fusion molecules by taking advantage of natural receptor-ligand interactions.
Abbreviations
| SEM | = | standard estimation of mean |
| SD | = | standard deviation |
| IP | = | immunoprecipitation |
| PBS | = | phosphate buffered saline |
| HUVEC | = | human umbilical vein endothelial cells |
| IgG | = | immunoglobulin G |
| ADSC | = | adipose-derived stem cells |
| ECFC | = | endothelial colony-forming cells |
| SEC-MALS | = | size exclusion chromatography-multi angle light scattering |
| MFI | = | median fluorescent intensity |
| FITC | = | fluorescein isothiocyanate |
| PFA | = | paraformaldehyde |
| HRP | = | horseradish peroxidase |
Introduction
The disruption of tumor growth by targeting multiple signaling pathways with monoclonal antibodies (mAbs) has proven effective in the treatment of cancer. In the past decade, notable progress has been made in the engineering and development of dual-targeting, bispecific antibodies (bsAbs) as therapeutics.Citation1 Since the approval of the first bsAb, catumaxomab, in 2009, 24 bsAbs have entered clinical evaluation in various indications.Citation2,3 However, compared with conventional mAb development, creating an engineered bsAb with adequate developability and manufacturability poses substantial challenges, usually leading to prolonged, iterative development cycles.Citation4-7 Many of the issues associated with the development of bsAbs are caused by the instability of the single chain Fv format (scFv), a common building block for these molecules.Citation6,8-10 Recently, platform approaches aimed at creating bivalent bsAbs closely resembling the traditional “Y”-shaped immunoglobulin (IgG) architecture with the potential for improved druggability have been reported.Citation11-15 Although development timelines are much shorter for these newly described platforms, they still require substantial engineering efforts, additional downstream steps for proper assembly or complicated stable cell line generation process. In addition, these formats yield monovalent binding moieties, which may potentially affect function due to loss of avidity. As an alternative approach, the building blocks for constructing dual-targeting antibodies could also originate from non-antibody natural proteins. Immunocytokines, for example, are a class of bi-functional molecules that fuse cytokines, often IL-2, to an IgG or antibody fragment.Citation16 Since many natural ligand-receptor interactions involved in cell growth and survival pathways are druggable targets, development of a bi-functional tetravalent antibody-like molecule through fusion of the native ligand binding domain of a receptor to an IgG is an attractive alternative strategy to traditional bsAb formats.
Vascular endothelial growth factors (VEGFs) are a family of closely related proteins with human VEGF-A165 (hereafter referred to as VEGF) being the most abundant and active isoform.Citation17 Through binding to VEGFR2, VEGF regulates critical steps in angiogenesis, from endothelial cell proliferation to migration. With the approval of bevacizumab in 2004, VEGF became a clinically validated target in oncology.Citation18-20 VEGFR1 exists in either membrane-bound or soluble (sVEGFR1) form with higher affinity for VEGF than VEGFR2.Citation21 sVEGFR1 is believed to function as a “scavenger” to sequester circulating VEGF, thereby modulating the strength of the signal from the angiogenic VEGF-VEGFR2 axis. Since the isolated domain 2 (D2) of VEGFR1 can bind to VEGF with nanomolar affinity as a monomer,Citation22 it was selected as the “building block” for construction of a bi-functional molecule. A similar concept has been previously validated in a monotherapeutic molecule aflibercept (VEGF trap) ― a VEGFR1 D2-VEGFR2-D3 chimeric fusion to the N-terminus of IgG Fc domain.Citation23
Insulin-like growth factor-I receptor (IGF-IR) plays a critical role in the development, maintenance and progression of many solid and hematopoietic malignancies.Citation24,25 IGF-IR also confers resistance to many cytotoxic, hormonal, and targeted therapies.Citation24,25 Preclinically, mAbs targeting IGF-IR that block binding of the ligands (IGF-I, IGF-II) cause down-regulation of the receptor, and have demonstrated anti-tumor activities in monotherapy and in combination with cytotoxic, radiation and targeted therapies.Citation4,24,26 To date, clinical activity of anti-IGF-IR antibodies in oncology has been modest. Therefore, combination with another targeted agent may increase the clinical response to anti-IGF-IR therapy. Recently, IGF-IR has been implicated in the regulation of angiogenesis and lymphangiogenesis.Citation27 Blockade of IGF-IR leads to inhibition of VEGF production, reduction in angiogenesis, and enhancement of anti-VEGF therapy.Citation27-29 Moreover, hypoxia results in IGF-II upregulation, which may potentially mediate tumor escape from anti-VEGF therapy.Citation30,31 These data suggest the combination of anti-IGF-1R and anti-VEGF treatments may lead to improved anti-tumor activity.
We describe herein the design and development of a novel bi-functional antibody that fuses a receptor domain to an IgG and simultaneously targets IGF-IR and VEGF. The bi-AbCap presented in this report possesses excellent developability, good thermal and physical stability, and acceptable clearance in vivo without extensive engineering. In addition to blocking the interaction of ligand and receptor at both targets, removal of VEGF by an intriguing “capture-for-degradation” mechanism correlates to superior anti-tumor activity in vivo compared to monotherapies or combinations of single agents. Our case study suggests the approach of appending a natural protein binding domain, like VEGFR1 D2, to an antibody as a practical alternative to the development of traditional bsAbs.
Results
Design and construction
Many ligands and receptors interact with affinities in the nanomolar range. The extra-cellular domain (ECD) of a receptor, or a functional portion of the ECD, has been used successfully as a “ligand trap” for therapeutic purposes.Citation21,22,32 Therefore, we decided to explore the feasibility of building an IgG-based, bi-functional molecule using a receptor domain with high affinity for its ligand as a building block to provide a second binding specificity to a selected IgG. To test the hypothesis that the efficacy of anti-IGF-IR and anti-VEGF therapies could be further enhanced by co-targeting both pathways, we combined an anti-IGF-IR antibody with a VEGF binding domain (D2) of human VEGFR1 in a novel format, the bi-functional antibody receptor domain fusion molecule with ligand capture (bi-AbCap). A human monoclonal IgG1 antibody targeting IGF-IR IR mAb (IMC-A12),Citation24 previously derived from a phage display library, was selected as the backbone for the Bi-AbCap (). The mechanisms of action for the IR mAb include receptor binding, blockade of the receptor/ligand interaction, and receptor internalization and degradation. VEGF binds to VEGFR1 and sVEGFR1 with ~10-fold higher affinity than to VEGFR2.Citation33-35 Based on previous reports of the tight association between VEGFR1 D2 and VEGF (Kd = 1.4 nM as a monomer),Citation22,23 we selected D2 (residues 129–229, calculated MW = 11.5 kDa) as the receptor domain partner to target VEGF and fused it to the carboxy-terminus (C-terminus) of the heavy chain (Hc) of IR mAb via a G4SG4 linker (). The resulting bi-functional molecule was named ID2. A D2I bi-AbCap, with D2 fused to the amino-terminus (N-terminus) of the IR mAb light chain (Lc), was also made and tested in vitro in an IGF-I stimulated IGF-IR phosphorylation assay using BxPC-3 human pancreatic cancer cells (Fig. S1A and S1C). Unlike ID2, D2I could not potently neutralize phosphorylation of IGF-IR in a dose-dependent manner as ID2 (Fig. S1B and S1C), possibly due to occlusion of antigen binding by the N-terminal VEGFR1 D2. Therefore, only the C-terminal fusion ID2 was used for further analysis. A monospecific FcD2 fusion molecule was also created as anti VEGF control ().
Figure 1. Design and molecular structure of bi-functional antibody receptor domain fusion molecule with VEGF capture (bi-AbCap). (A) Cartoon illustration of the designed bi-AbCap fusion molecule. Orange: Anti IGF-IR IgG backbone, IR mAb; Purple: polypeptide linker; Green: extracellular domain 2 of human VEGFR1 (D2). An FcD2 control molecule was made containing the regions of the hinge, Fc, linker and D2. (B) SDS-PAGE of IR mAb, FcD2 and bi-AbCap ID2. (C) Top: linker sequence: G4SG4; Bottom: the D2 domain used for fusion encompassing amino acids 129–229 of hVEGFR1. (D) ID2 elutes as a mono-disperse peak by size exclusion chromatograpy (SEC). (E) ID2 displays 3 melting temperatures (Tm) when the thermal stability of ID2 measured by differential scanning calorimetry (DSC). Tm1 = 64.4 ± 0.0°C, Tm2 = 76.0 ± 0.0°C, Tm3 = 82.7 ± 0.1°C. (F) IR mAb displays 3 Tms by DSC: Tm1 = 72.2 ± 0.1°C, Tm2 = 76.8 ± 0.0°C, Tm3 = 83.2 ± 0.1°C.
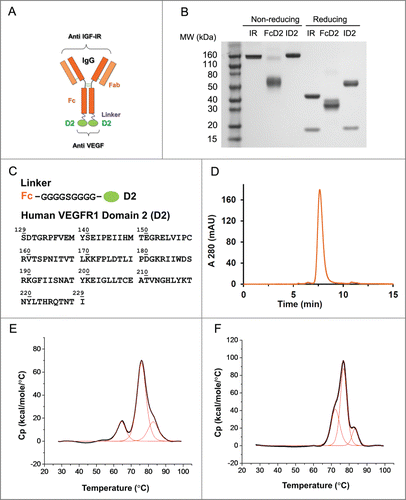
Developability and stability
To investigate whether the bi-AbCap is suitable for downstream development, various studies were carried out to assess the developability and stability of the bi-functional molecule. The average expression level of ID2 from stably transfected Chinese hamster ovary (CHO) cells was 1,127 ± 155 mg/L (n = 2), similar to the expression of a typical IgG1. After purification over a protein A column, ID2 eluted as a single peak upon size exclusion chromatography (SEC), with ~97% of the molecule in monomeric form (), and showed little evidence of aggregation. Assessment of processability indicated that bi-AbCap could be purified using a standard commercial platform purification process.
ID2 also demonstrated thermal stability similar to a typical IgG1 (). Upon analysis by differential scanning calorimetry (DSC), the D2 domain showed a melting temperature of around 64 °C. The melting temperatures for the CH2 and Fab domains overlapped in the region of 76 °C, while the CH3 domain unfolded at around 83°C (). Thus, the thermal stability of the IgG portion of ID2 is similar to that of IR mAb (). Furthermore, upon incubation for up to 7 d at 37 °C in the presence of 10% mouse serum, the binding activities of ID2 to IGF-IR and VEGF were completely retained, indicating good structural stability of the molecule. (Fig. S2).
The stability of the bi-AbCap was also evaluated at high protein concentration. ID2 was concentrated from 9.2 to 67 mg/ml in PBS and tested for insoluble and soluble aggregates by filtration and SEC, respectively. At both concentrations, all of the ID2 was recovered after filtration (0.1 µm) (). SEC analysis of ID2 at 9.2 and 67.0 mg/mL showed similar profiles with no evidence of soluble aggregates ().
Table 1. Summary of physical stability of ID2 at high concentration
Anti-IGF-IR activity
To confirm that C-terminal fusion of VEGFR1 D2 does not interfere with the activities of the parental anti-IGF-IR mAb, ID2 was tested for function in IGF-IR binding ELISA and in cell-based assays. ID2 bound to IGF-IR ECD with an EC50 of 0.30 nM by ELISA and a Kd of 0.22 nM by surface plasmon resonance (SPR, and ). ID2 also was shown to inhibit the interaction of IGF-I and IGF-IR in a blocking assay with an IC50 of 1.65 nM (). Similar results were observed with IR mAb ( and ). In BxPC-3 and MCF-7 tumor cells, pre-incubation with 100 nM ID2 inhibited IGF-I-stimulated phosphorylation of IGF-IR, ERK1/2, and AKT. The level of reduction was similar to that observed after pre-incubation with 100 nM IR mAb (). In addition, treatment with ID2 or IR mAb caused a slight reduction in the level of total IGF-IR in these cells compared with control treatment, suggesting down regulation of the receptor (). The ability of ID2 to internalize and degrade IGF-IR was confirmed by ELISA using MCF-7 cells (). Time-dependent down-modulation of IGF-IR was determined using total cell lysates after treatment with ID2, IR mAb, or IgG control. During the first 4 hours, total IGF-IR decreased rapidly in the presence of ID2 and 77% of the receptor disappeared (). From 4 to 24 hours, the amount of IGF-IR decreased by an additional 6% after treatment with ID2 (). Overall, the rate of IGF-IR down-modulation by ID2 was similar to that observed with IR mAb (). Treatment with ID2 also inhibited the growth of MCF-7 cells stimulated with IGF-I with an IC50 of 5.1 nM, comparable to that observed after treatment with IR mAb (). These data indicated that the bi-AbCap retained the in vitro activities of IR mAb.
Figure 2. Binding to IGF-IR, VEGF and evidence for co-engagement of both targets by ELISA. (A) ID2 (orange), IR mAb (blue) and human IgG control (gray) binding to human IGF-IR-Fc. (B) ID2 (orange), FcD2 (green) and human IgG control (gray) binding to human VEGF. (C) ID2 (orange), IR mAb (blue) and FcD2 (green) binding to both human IGF-IR-Fc (coated) and human VEGF (detected). Each panel is a representative experiment of at least 3 repeated measurements. The graph is plotted as mean ± SEM (n = 2 ).
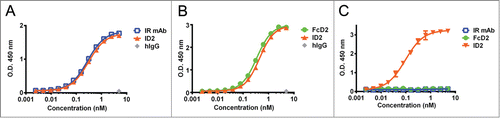
Figure 3. Inhibition of IGF-IR-mediated tumor cell signaling and functions by ID2. (A) 100 nM ID2 inhibits IGF-I-induced phosphorylation of IGF-IR, downstream AKT and ERK1/2 in BxPC-3 cells as assessed by immunoblotting analysis. IR mAb and FcD2 were used as controls. (B) 100 nM of ID2 inhibits IGF-I-induced phosphorylation of IGF-IR, downstream AKT and ERK1/2 in MCF-7 cells in immunoblotting analysis. IR mAb and FcD2 were used as controls. (C) Down regulation of surface IGF-IR on MCF-7 cells when treated with ID2, control IR mAb and control human IgG is measured at 0, 1, 4, 8 and 24 hours by IGF-IR electro-chemiluminescence (ECL) assay. (D) ID2 potently inhibits IGF-I induced MCF-7 viability in a dose dependent manner in a CellTiter Glo assay. The error bar represents the SEM from each triplicate measurement.
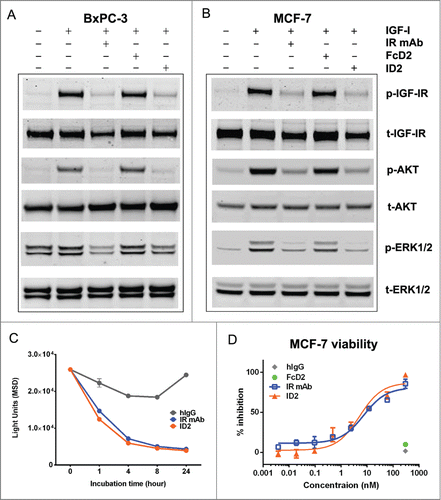
Table 2. Summary of ELISA binding, affinity (SPR), and ELISA blocking activities
Anti-VEGF activity
The anti-VEGF activity of the bi-AbCap was further evaluated using in vitro assays. ID2 bound to VEGF with an EC50 of 0.45 nM, as determined by ELISA and a Kd of 0.18 nM, as determined by SPR ( and ). ID2 also blocked the interaction of VEGF and VEGFR2 with an IC50 of 2.60 nM in a blocking ELISA format (). Similar results were obtained using a control, FcD2 ( and ). In cell signaling assays, 100 nM ID2 led to potent inhibition of the phosphorylation of VEGFR2, ERK1/2, and AKT in porcine aortic endothelial (PAE) cells overexpressing human VEGFR2/KDR (kinase insert domain receptor) (PAE/KDR) (). This level of inhibition was similar to that observed with FcD2.
Figure 4. Inhibition of VEGF-mediated endothelial cell signaling and functions by ID2. (A) 100 nM ID2 inhibits VEGF-induced phosphorylation of VEGFR2, downstream AKT and ERK1/2 in PAE/KDR cells as assessed by immunoblotting analysis. IR mAb and FcD2 were used as controls. (B) In an Oris cell migration assay, PAE/KDR cells stimulated with 100 ng/ml VEGF were treated with 100 nM ID2, IR mAb, or FcD2 for 20 hours. The fluorescence intensity of migrated cells in relative fluorescence units (RFU) was measured. ID2 significantly reduced the migration compared to VEGF and IR mAb controls (p = 0.002 and p = 0.003, respectively, one way ANOVA). (C) ID2 inhibits VEGF stimulated cord formation in an ADSC/ECFC co-culture system. The total tube area for each treatment was calculated. ID2 significantly reduced the total tube area compared with VEGF only and IR mAb controls (p < 0.0001 and p < 0.0001, respectively, one way ANOVA). (D) ID2 inhibits human VEGF induced HUVEC viability in a dose dependent manner in a CellTiter Glo assay. The error bar from panels B, C and D represents the SEM from each triplicate measurement.
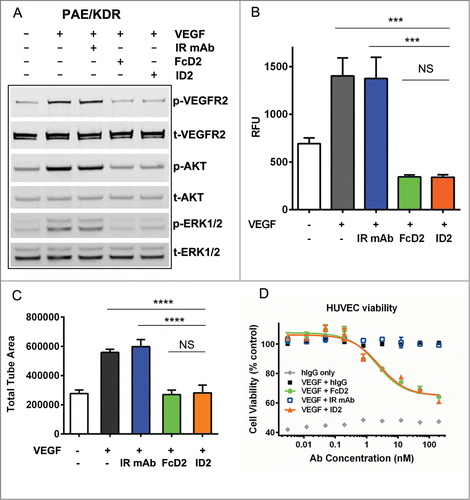
Since endothelial cell migration is an essential part of angiogenesis, the anti-migratory activity of ID2 was evaluated in an endothelial cell migration assay (). At 100 nM, ID2 significantly reduced the migration of PAE/KDR cells in response to stimulation with VEGF. This inhibitory effect was also observed with FcD2, but not with IR mAb ().
To further assess the effect of VEGF blockade by the D2 arm of ID2, an ADSC/ECFC co-culture cord formation assayCitation36 was performed. Treatment of cords with ID2 and FcD2 for 3–4 d following VEGF induction was shown to decrease total tube area, while IR mAb alone had no effect on total tube area ().
In addition, in a human umbilical vein endothelial cell (HUVEC) viability assay, ID2 bi-AbCap inhibited cell growth stimulated by VEGF to the same extent as FcD2. IC50s of HUVEC growth inhibition were 2.5 nM for ID2 and 2.1 nM for FcD2 (). In conclusion, the D2 arm of the bi-AbCap demonstrated robust blockade of multiple processes involved in VEGF-stimulated angiogenesis in vitro.
It was reported previously that, unlike the high molecular weight oligomers formed by the binding of bevacizumab to VEGF, the VEGF trap molecule, constructed by fusing VEGFR1 D2 and VEGFR2 D3 to the N-term of the IgG Fc domain assembles as a 1:1 stoichiometric complex with the VEGF dimer.Citation37 Analysis of binding stoichiometry using SEC-MALS suggests that ID2 predominantly forms a 1:1 ratio with the VEGF dimer, showing minimal formation of aggregated oligomers (Fig. S3). Therefore, it is expected that the VEGF-bound bi-AbCap molecule would be less likely to form complexes with immunogenic potential.
A unique mechanism – targeting VEGF for degradation
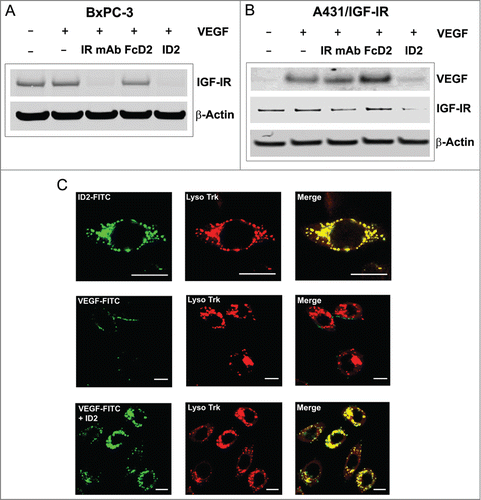
Since both tumorigenesis and angiogenesis contribute to tumor development, a therapeutic agent like ID2 has the potential to block both pathways simultaneously, and thereby inhibit tumor growth as effectively and perhaps more potently than the combination of 2 individual blocking antibodies. To further characterize the unique properties of ID2, we first verified the ability of this bi-AbCap to simultaneously engage and crosslink both IGF-IR and VEGF targets. In a dual binding ELISA, IGF-IR was coated onto a plate followed by the incubation with ID2, FcD2 or IR mAb. After detection using VEGF and a biotinylated anti-VEGF antibody, only ID2 was found to engage both IGF-IR and VEGF in a dose-dependent manner ().
Based on the bi-AbCap design, once the IR mAb portion of the molecule is engaged with IGF-IR on the surface of tumor cells, it is tempting to speculate that ID2 could provide enhanced inhibition of tumor growth through sequestration and internalization of VEGF. As suggested previously, down regulation of IGF-IR on the cell surface can be mediated by IR mAb and ID2 (). The ability of ID2 to reduce cell surface IGF-IR level was further confirmed by immunoblotting analysis. Treatment with either ID2 or IR mAb significantly reduced the level of total IGF-IR on BxPC-3 cells after 16 hours of incubation (). To test the hypothesis that the ID2 bi-AbCap is capable of inducing co-degradation of IGF-IR and VEGF simultaneously as a unique mechanism of action, a VEGF degradation assay was developed. A431 cells overexpressing IGF-IR (A431/IGF-IR) were incubated with exogenous VEGF and ID2 or control molecules. VEGF in cell culture supernatant, and IGF-IR from cell lysates, were detected by immunoblotting analysis. As expected, IGF-IR level after ID2 treatment was reduced compared to treatment with FcD2 and un-treated controls (). Interestingly, ID2 treatment also dramatically decreased the level of VEGF in the cell culture supernatant, while the VEGF level in control samples remained unchanged or slightly increased upon treatment with IR mAb or FcD2, respectively (). Furthermore, confocal immunofluorescence microscopy visualization showed that co-localization of FITC-labeled VEGF within the lysosomal compartment in BxPC-3 cells was dependent on the presence of ID2 (). At 37 °C, FITC-labeled ID2 showed a predominantly intracellular distribution which co-localized with a lysosome specific dye after incubation for 2 hours (, top). However, FITC-labeled VEGF alone only weakly stained along the edge of the cell, showing no overlap with lysosome staining and suggesting possible low-level binding to VEGFR1 on the cell surface (, middle). Upon co-incubation with un-labeled ID2, FITC-labeled VEGF shifted intracellularly and superimposed completely with the lysosome staining (, bottom).
Figure 5. Evidence for internalization and degradation of both IGF-IR and VEGF by ID2 in vitro. (A) In BxPC-3 cells, ID2 at 20 nM induces IGF-IR internalization and degradation in the presence of 100 ng/mL VEGF by immunoblotting. IR mAb and FcD2 were used as controls. (B) In A431/IGF-IR cells, ID2 at 10 nM induces both surface IGF-IR and supernatant VEGF internalization/degradation in the presence of 400 ng/mL (10 nM calculated as a dimer) VEGF by immunoblotting. IR mAb and FcD2 were used as controls. (C) Fluorescence confocal microscopy analysis on the delivery of ID2 and VEGF to the lysosome in BxPC-3 cells: (top row) the co-localization of FITC-labeled ID2 with lyso tracker; (middle and bottom row) the co-localization of FITC labeled VEGF with the lysosome is dependent on ID2 treatment. Scale bar = 20 μm.
A series of live cell imaging experiments were performed to further validate the trafficking and delivery of ID2 and VEGF to the lysosomes in A431/IGF-IR cells. First, a pH-activated dye (pHrodo™) conjugated to an anti-human IgG FAB antibody was used as a surrogate for detecting the delivery of antibody to the lysosome. Following a 20-hour incubation, a significant increase in the signal from pHrodo dye was evident for both IR mAb and ID2 compared with control human IgG group (Fig. S4A). Next, treatment with human IgG control, IR mAb or FcD2 caused only minimal internalization of VEGF labeled with Alexa-647, whereas ID2 was found to mediate efficient internalization and trafficking of labeled VEGF (Fig. S4B). Furthermore, high resolution time-lapse confocal microscopy revealed coincident localization of ID2 with VEGF on LAMP1-positive endosomes (Fig. S4C, representative still image), after A431/IGF-IR cells transfected with the late-endosomal marker LAMP1 conjugated to mCherry were incubated with Alexa-488 labeled ID2 along with VEGF-Alexa-647, demonstrating that the internalized ID2-VEGF complex was indeed targeted to the lysosome.
Taken together, cell-based degradation and imaging studies strongly suggest a unique mechanism of action for the bi-AbCap. Both IGF-IR on tumor cell surface and VEGF from the tumor environment are directed to the lysosomal degradation pathway by covalently linking IGF-IR and VEGF targeting moieties in the bi-AbCap format.
Inhibition of tumor growth in vivo
To compare the efficacies of ID2, IR mAb and FcD2, we examined the activity of the molecules in vivo in human tumor xenograft models. These initial doses and schedule (3 times per week) were selected to achieve the maximal efficacy for each treatment. In addition to IR mAb and FcD2, a rat anti-mouse VEGFR2 antibody DC101,Citation38 was also used as an anti-angiogenic monotherapy control. In both MiaPaCa-2 pancreatic and HT-29 colorectal cancer models, the monospecific molecules IR mAb, FcD2 and DC101 exhibited significant tumor growth inhibition (31–66%) compared to saline control (). Moreover, ID2 at 30 mg/kg demonstrated significantly better suppression of tumor growth compared to IR mAb (p < 0.0001 in both models), FcD2 (p < 0.0001 in MiaPaCa-2) and DC101 (p < 0.0001 in MiaPaCa-2; p = 0.04 in HT-29) (). Thus, ID2 demonstrated improved anti-tumor activity compared to monotherapy in vivo.
Figure 6. ID2 inhibits tumor growth in multiple mouse xenograft models and demonstrates superior anti-tumor activity compared to the IR mAb/FcD2 combination. Tumor bearing mice were treated with controls and ID2 by intraperitoneal injection 3 times per week. The figure shows tumor volume (in mm3) as a function of treatment time (in days). The models tested were: (A) MiaPaCa-2; (B) HT-29; (C) Caki-1; (D) Colo-205. Tumor volume is plotted as mean ± SEM (n = 12). For statistical analysis, RM ANOVA was used through the last day of each study to compare the tumor growth between ID2 and other treatments. Levels of statistical significance are indicated as: *p = 0.01–0.05; ***p = 0.0001–0.001; ****p < 0.0001.
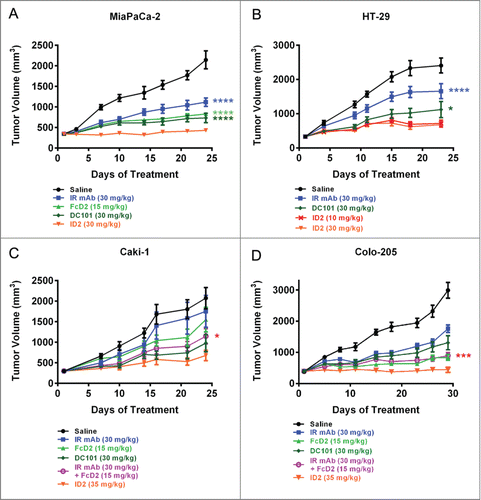
Since treatment with ID2 under in vitro setting induced degradation of both IGF-IR and VEGF, we examined whether this distinct mechanism of action could potentially lead to more potent efficacy in vivo. To minimize the potential impact of different molecular weights on dosing levels, all reagents were administered at equimolar doses 3 times per week (). In both Caki-1 and Colo-205 xenograft models, ID2 improved the anti-tumor efficacy over the monospecific IR mAb or FcD2 () as expected. More impressively, ID2 exhibited superior anti-tumor activity over the combination of IR mAb and FcD2 in these tumor models (p = 0.0254 in Caki-1; p = 0.0002 in Colo-205) (). The in vivo data indicate that ID2 achieves superior anti-tumor activity in vivo compared to either monospecific or combination therapy. It is likely that the additional activity observed with ID2 may be a result of the unique co-degradation mechanism that is absent when IR mAb and FcD2 are combined.
Pharmacokinetic properties of ID2 and its derivative
Adequate stability in vivo is critical for the development of bi-specific or bi-functional molecule, in order to enable standard (e.g., once-weekly) dosing and schedule. Pharmacokinetic (PK) studies were performed in order to better understand the in vivo stability characteristics of the bi-AbCap ID2. CD-1 mice were dosed intravenously with 30 mg/kg ID2, 30 mg/kg IR mAb or 15 mg/kg FcD2 and serum concentrations of total IgG for ID2 and IR mAb or Fc for FcD2 were determined over a 2 week period ( and ). In this study, ID2 was shown to possess a shorter half-life (T1/2) (66 vs. 141 hours) and 3-fold faster clearance than the IR mAb ( and ). The PK parameters for FcD2 in a separate study with faster clearance but longer T1/2 showed much smaller differences from ID2 (). Since ID2 fully cross-reacts with mouse IGF-IR and mouse VEGF (), it is possible that ID2 is susceptible to target-mediated clearance. The more rapid clearance of ID2 relative to IR mAb could be attributed to unknown mechanisms, such as non-specific clearance, clearance specific to VEGF targeting, or a combination of both processes. The inability to saturate the clearance mechanism in the case of ID2 at higher doses (data not shown) does suggest at least some contribution of non-specific clearance.
Figure 7. In vivo stability of ID2. (A) Pharmacokinetic assessment of serum concentrations of ID2 and IR mAb as a function time (in hours), following 30 mg/kg intraveneous administration in CD-1 mice (n = 3). Total IgG was used for determining the serum concentrations. The serum concentrations were plotted as mean ± SD. (B) Dose frequency study in a Colo-205 mouse xenograft model. 2.5 mg/kg (orange line), 12.5 mg/kg (blue line) and 35 mg/kg (purple line) of I3D2 (an engineered variant of ID2) were dosed intraperitoneally once weekly (light colored line) and twice weekly (dark colored line) schedules. Tumor volumes are plotted as mean ± SEM (n = 12).
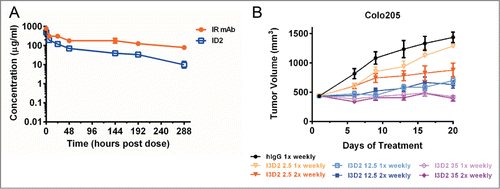
Table 3. Summary of mouse PK data for bi-AbCap ID2, FcD2 and IR mAb
A derivative of ID2, I3D2, with a few amino acid changes both at the end of λLc Citation39 and in Hc-D2 region was made to improve chemical stability (Table S1) under stress conditions and demonstrated similar in vitro and in vivo activity compared to ID2 (data not shown). Although I3D2 displayed slightly improved pharmacokinetics in CD-1 mice in comparison to ID2, the half-life is still around 3 d (Table S2).
To further validate whether our bi-AbCap with once weekly dosing would achieve maximal efficacy, a comparison of dose regimens was performed. The Colo-205 model was selected because ID2 demonstrated the most potent tumor inhibition in that model and it would, therefore, provide a wide window for comparison of efficacy. I3D2 at low (2.5 mg/kg), intermediate (12.5 mg/kg) or high (35 mg/kg) dose was injected on once weekly and twice weekly schedules. I3D2 demonstrated dose dependent inhibition of tumor growth across the 3 dose levels. Given the fact that I3D2 has a T1/2 of around 3 days, it is surprising that, at both high and intermediate dose levels, no difference in inhibition of tumor growth was observed with the once weekly dosing regimen (p = 0.2059 for 12.5 mg/kg and p = 0.7537 for 35 mg/kg) compared to twice weekly dosing (). However, maximal inhibition of tumor growth was only observed at 35 mg/kg dosed once weekly ().
Collectively, in spite of the short half-life, the data indicate that the bi-AbCap could be administered once a week at 35 mg/kg to reach maximal anti-tumor efficacy in a mouse xenograft model tested.
In vivo mechanism of action for tumor growth inhibition
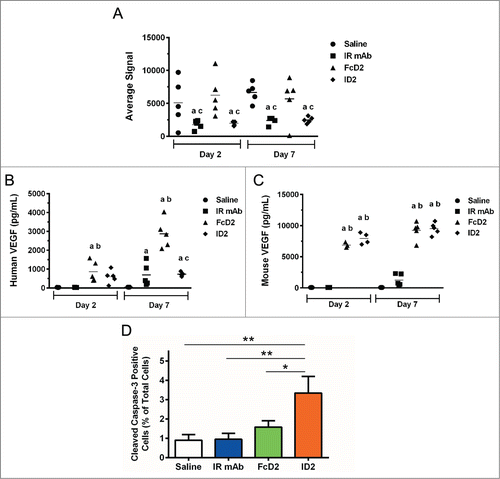
This potent inhibition of both tumor growth and tumor angiogenesis was confirmed by mechanism of action studies using the same Colo-205 model. Treatment with saline, IR mAb, FcD2 or ID2 was given on Day 1, 3 and 5. Tumor tissues and blood were collected and processed on Day 2 and 7. Both ID2 and IR mAb treatments significantly reduced tumor IGF-IR level by electrochemiluminescent assay compared to treatment with saline or FcD2 (). While FcD2 caused an elevation of circulating human VEGF, possibly due to the build-up of FcD2-hVEGF (), compared to saline or IR mAb treated control groups, this effect was attenuated in plasma from mice treated with ID2 after Day 7 (). However, circulating mouse VEGF from ID2 treated group after Day 2 or 7 remained at a similar elevated level as that from FcD2 treated group (). These findings suggest the possibility of more efficient human VEGF degradation mediated by ID2 in the circulation and/or vicinity of tumor tissue in vivo (see discussion and Fig. S6). Moreover, a significant increase in percentage of cells with positive cleaved caspase-3 staining was observed on Day 2 in the tumor treated with ID2 in comparison with the tumors treated with saline, IR mAb or FcD2 (). As apoptosis mediated by caspase-3 could be activated not only by IGF-IR inhibition, but also by anti-VEGF treatment and, therefore, was associated with vessel regression,Citation40,41 our data are consistent with the previous finding and demonstrate a higher level of apoptosis upon bi-AbCap treatment than IR mAb or FcD2 alone. In short, the mechanism of action studies not only confirmed the capability of bi-AbCap to block both targets, but also suggested potential mechanisms, such as co-degradation and increased apoptosis, for its superior activity.
Figure 8. In vivo mechanism of action in Colo-205 xenograft model. (A) Average signal of total human IGF-IR from lysates of excised tumor, (B) human VEGF concentration (pg/ml) from mouse plasma and (C) Mouse VEGF concentration (pg/ml) from mouse plasma after 2- and 7-day treatments with saline, IR mAb, FcD2 and ID2 were determined by electrochemiluminescent assay. Significant differences in mean (p < 0.05, n = 5) are indicated (a vs Saline; b vs IR mAb; c vs FcD2). (D) Percentage of nuclei with positive immunohistochemistry staining for cleaved caspase-3 from representative tumor tissue after 2-day treatment was compared and plotted as mean ± SEM (n = 5). ID2 treated group had significantly increased cleaved caspase-3 activity compared to saline control (p = 0.0035, one way ANOVA), IR mAb (p = 0.0041, one way ANOVA) and FcD2 (p = 0.0254, one way ANOVA). All charts were generated and statistical analyses were performed with SigmaPlot or Graphpad Prism 6.
Discussion
In this report, we describe the development of a novel bi-functional molecule created by fusing a receptor domain from human VEGFR1 to a mAb directed against IGF-IR. In contrast to the scFv-based format used to make traditional tetravalent bsAbs, which require extensive de novo screening or subsequent engineering to curb soluble aggregation and improve thermal stability,Citation8,10 there are advantages conferred by the unique features of this design. The fusion of a single, stable domain to an IgG allows the construction of a bi-functional molecule with minimal engineering. In fact, ID2 demonstrated impressive solubility with no sign of aggregation at up to 67 mg/mL. In addition, the unmodified D2 domain bound VEGFA-165 with sufficient affinity to demonstrate biological activity in vitro and in vivo. The bivalent VEGFR1 D2 domain in ID2 blocked the VEGF-VEGFR2 interaction with an IC50 of 2.6 nM, similar to the blocking activity of the anti-VEGF antibody bevacizumab (IC50 of 1.4 nM). No affinity maturation was required for the D2 arm of the bi-AbCap. Besides binding to VEGFA-165, ID2 also binds to VEGF-B and PlGF with similar affinity to VEGFA-165 (data not shown). The biological relevance of VEGF-B and PlGF blockade was not investigated in our study.
The limited clinical response observed with antibodies directed against IGF-IR and the recent preclinical evidence suggesting a regulatory role for IGF-IR in VEGF expression, angiogenesis, and lymphangiogenesis prompted us to seek enhancement of current therapeutic options through a dual targeting reagent.Citation26-29 In addition to direct blocking activity, the bi-AbCap promotes co-internalization and degradation of both targets through the lysosomal pathway in vitro. The ability of ID2 to remove exogenous VEGF from A431/IGF-IR cell culture supernatant is possibly related to the superiority of bi-AbCap activity to the combination therapy observed in vivo. Furthermore, similar to reported observations from patients treated with bevacizumab,Citation42 both human and mouse VEGFs in plasma from Colo-205 xenograft mice treated with either ID2 or FcD2 were elevated compared with saline control group (). Interestingly, only the plasma level of human VEGF from ID2-treated animals was significantly lower than that from the group treated with FcD2 (), a scenario reminiscent of the slight increase of supernatant VEGF from FcD2 and the decrease of VEGF from the ID2 treated group observed in the cell based study ().
The reason for this discrepancy is unknown, but can be postulated. One explanation is that ID2 can degrade human VEGF more efficiently in the tumor vicinity because of higher expression of tumor IGF-IR and higher local concentration of human VEGF as a result of direct secretion from tumor cells. IGF-IR mRNA levels in the TCGA (The Cancer Genome Atlas) RNA-seq datasets that contained 6,943 samples representing 21 tumor types were evaluated to understand the prevalence of IGF-IR expression in normal and tumor tissues (Fig. S5). We previously confirmed that there was a strong correlation between IGF-IR mRNA and protein (data not shown). In normal tissues, the highest levels of IGF-IR expression were observed in breast, prostate, kidney, endometrium and thyroid gland. IGF-IR is overexpressed in multiple tumor types including squamous cell carcinoma of the lung, breast, prostate cancer and sarcoma. Some of these tumors display strikingly high IGF-IR expression. Therefore, while it is plausible that IGF-IR in certain organs, for example, kidney and mammary gland, may contribute to ID2 mechanism of action, the pattern of IGF-IR expression suggests that IGF-IR in the tumor compartment may be primarily responsible for the “sink” effect. As internalization and degradation are dependent primarily on IGF-IR expression on tumor cells, it is possible that ID2, targeted to the tumor by binding to IGF-IR, could more efficiently capture and degrade the human VEGF concentrated around the tumor tissue. As the circulating concentration of human VEGF is lower than that of mouse VEGF in animals treated with FcD2 (), a decrease in human VEGF near tumor tissue might result in a more pronounced reduction in plasma.
The other possible reason for the discrepancy between human and mouse VEGF level changes upon ID2 treatment could be attributed to the difference in internalization/degradation rate between human and mouse VEGF. Using the described degradation assay (), the kinetics of biotinylated human VEGF and biotinylated mouse VEGF degradation upon ID2 treatment was compared during a 24-hour period. Interestingly, human VEGF was internalized and degraded much more quickly between 2–24 hours than mouse VEGF (Fig. S6). It is unknown, however, why the rate of degradation is inconsistent between mouse and human VEGFs, given that the EC50s of ID2 binding to the biotinylated human and mouse VEGF are identical (data not shown).
Since human VEGF binds equally well as mouse VEGF to mouse VEGFR2 (Fig. S7) and the blockade of human VEGF by bevacizumab (does not bind to mouse VEGF) was effective to reduce tumor angiogenesis in Colo-205 mouse xenograft model, Citation43 the observed decrease in human VEGF in ID2-treated, but not FcD2-treated group does have its therapeutic relevance and may represent the unique evidence for bi-AbCap to modulate angiogenesis more effectively around the tumor in these mouse xenograft models. Further in vivo mechanism action studies are warranted to validate the therapeutic relevance of this “trap-for-degradation” mechanism by bi-AbCap.
It was reported that the “sweeping antibodies” against soluble human IL-6R (hsIL-6R) via the combination of pH dependent target binding engineering and enhanced FcRn binding at neutral pH induce a 50- to 1000-fold decrease of plasma hsIL-6R concentration compared with a conventional antibody.Citation44 In contrast to the “sweeping antibody,” our bi-AbCap differs in the following ways. First, bi-AbCap relies on IGF-IR as a membrane anchoring receptor as opposed to more ubiquitously expressed FcRn for “sweeping antibody” to access the endosome-lysosome pathway. Second, bi-AbCap does not bind to VEGF in a pH-dependent manner. Third, bi-AbCap elevates mouse or human VEGF in circulation above the baseline level (Saline control group), but to a much lesser extent for human VEGF compared with FcD2 ( and 8C). Therefore, we postulate that the uniqueness of bi-AbCap design enables a more local VEGF clearance in a tumor environment preferably where higher tumor-than-normal tissue IGF-IR expression exists. This proposed “target-for-degradation” mechanism is likely the cause of the superior anti-tumor activity observed with the bi-AbCap, as the benefit of engaging 2 targets is maximized by physical linkage between IR mAb and D2.
Given the fact that IR mAb and ID2 display different PK profiles, it is possible that additional, as yet unidentified, VEGFR1 D2-dependent and independent mechanisms contribute to faster bi-AbCap clearance and shorter half-life. The cause for less than optimal PK profile for the control FcD2 is difficult to dissect, as the size (75 kDa) and format of the control are very different from a traditional IgG. However, in spite of the relatively rapid clearance observed for the bi-AbCap in mouse, our dosing frequency study established that a once weekly dosing schedule is sufficient to achieve maximal efficacy in a mouse model.
The concept of using receptor domains to generate chimeric molecules has been validated successfully in clinical studies or is still being tested as the mono-specific ligand traps, VEGF Trap or FGF Trap, respectively. These molecules were engineered by fusing corresponding receptor domains to the Fc portion of IgG at the N-terminus.Citation23,32 The idea of using a bispecific format to bind and eliminate VEGF through the internalization pathway of a membrane target was investigated previously.Citation8 A bsAb developed against PDGFRβ and VEGF was shown to internalize into human brain vascular pericytes and demonstrated activity similar to anti-VEGF treatment with bevacizumab in mouse models.Citation8 Our study is the first example offering direct evidence of VEGF degradation in tumor cells, both in vitro and in vivo, by a bi-functional molecule co-targeting VEGF and a cell surface receptor. The bi-AbCap demonstrates potent anti-tumor activity superior not only to anti IGF-IR or anti VEGF therapy, but also to the combination therapy, suggesting that targeting with a bi-specific/functional molecule is an advantageous strategy in achieving better efficacy than the combination under certain conditions where additional mechanism of action could be enabled. The bi-AbCap format represents an alternative approach to the creation of a dual targeting entity by exploiting the potential of a natural protein scaffold rather than the antibody fragments used traditionally in bi-specific antibody formats. As a result of this unique approach, minimal effort is required with respect to engineering and the developmental path is straightforward. The bi-AbCap demonstrates antibody-like developability and potent anti-tumor activity. Beyond the VEGF-VEGFR pathway, the fusion partner could originate from other receptor tyrosine kinase families or immune checkpoint receptors, thereby providing multiple opportunities for development of bi-functional IgG fusions. The bi-functional design, an IgG-receptor domain fusion, presented here represents a valuable alternative to traditional bispecific antibodies for the development of potent anti-tumor agents.
Materials and Methods
Design, construction and production
DNA encoding the extracellular domain 2 (D2) of human VEGFR1 encompassing residues 129–229 aa was amplified by PCR and cloned into mammalian expression constructs harboring IR mAb IgG, either before the N-terminus of IR mAb Lc via a linker GGGSGGGS – D2I, or after the C-terminus of IR mAb Hc via a linker GGGGSGGGG – ID2. The co-expression constructs were made by digesting corresponding Lc and Hc segments followed by re-ligation.
HEK 293-Freestyle cells (Life Technologies) transiently transfected with bi-AbCap expression constructs according to manufacturer's specifications or stably transfected CHO cells expressing ID2 were cultivated in suspension at 37°C in serum-free media. Cell culture fluid containing the bi-AbCaps was harvested after 6 d for transient and 2 weeks for stable CHO. MabSelect SuReTM Protein A (GE Healthcare) was used to purify the bi-AbCaps present in the harvested cell culture fluid from either transient HEK293 cells or stable CHO cells using an AKTA Explorer under the control of UNICORN 5.0 (GE Healthcare) as described previously.Citation45
Stability measurements
Differential Scanning Calorimetry (DSC). Thermal unfolding of ID2 was measured at 1.0 mg/mL in PBS using a MicroCal VP-Capillary DSC. The sample was scanned over the temperature range 20–95ºC with a scan rate of 60ºC/hour. The solutions were pressurized at about 60 psi in the capillaries during each scan. For each run, buffer/buffer scan was subtracted from buffer/protein scan and the thermogram was normalized for protein concentration. Baseline correction was performed and the midpoint of thermal denaturation (Tm) was obtained from the peak maximum.
Size Exclusion Chromatography. Samples (1 mg/mL, 100 μl) were analyzed on Agilent HPLC system using TSK-GEL G3000SW 7.8 mm × 30.0 mm column with a 20-minute run time and a flow rate of 1 ml/min (running buffer: 1× PBS at pH 7.4, 0.05% sodium azide). Data were analyzed using Chemstation software.
Size-exclusion chromatography-Multi angle light scattering (SEC-MALS). Samples were analyzed on an Agilent HPLC system equipped with a Wyatt MiniDawn Light Scattering detector, an Agilent Refractive Index detector, and an Agilent UV detector. Samples ran for 40 minutes on a Wyatt SEC column (7.8 × 300 mm, 300 Angstrom, Cat# 030S5G) with a flow rate of 0.4 ml/min (running buffer: 1× PBS at pH 7.4, 10% ethanol, 0.05% sodium azide). Absorbance at 280 nm, refractive index (RI) and light scattering (LS) were recorded. Data were analyzed using Astra 4.1 software.
Solubility study. Samples were concentrated sequentially from 9.2 mg/mL to 67 mg/mL using a 4 mL centrifugal 30MWCO filter device. Sample concentration was measure before and after 0.1 mm filtration and % recovery was calculated. Visual appearance of the solution was recorded and the percentage of soluble aggregates at low and high concentration were determined by SEC.
ELISA binding, blocking and surface plasmon resonance measurements
The general procedure for ELISA was as described. Citation5 For target binding assay, 100 ng/well human IGF-IR (R&D 391-GR-050), mouse IGF-IR (R&D 6630-GR-025/CF), human VEGFA-165 (R&D 293-VE-010), or mouse VEGF-164 (R&D 493-MV-025/CF) was immobilized and bound antibodies were detected by goat anti-human (Fab)'2 HRP conjugated antibody – for IGF-IR binding (Jackson ImmunoResearch 109–035–097) or anti-human Fc HRP conjugated antibody (Jackson ImmunoResearch 109–035–098) – for VEGF binding. To assess dual target binding, a 1:1 mixture (by volume) of serial diluted antibodies and 20 nM of VEGF was added to the plate coated with 100 ng/well IGF-IR. Bound antibody-VEGF complex was detected with a biotinylated anti-hVEGF antibody (R&D BAF293), followed by streptavidin HRP (Jackson ImmunoResearch 016–030–084). For the VEGF-VEGFR2 blocking ELISA, the pre-incubated mixture containing 4 nM human VEGF and varied amount of antibody was added to plates coated with 40 ng/well of VEGFR2 (R&D 357-KD). Bound VEGF was quantified as described in dual target binding. The IGF-IR blocking ELISA was performed following the method described previously.Citation24 For the SPR study, the binding kinetics of ID2 and parental controls to IGF-IR and VEGF were measured using a Biacore 2000 instrument. The data were evaluated using BIA Evaluation 2.0 (GE Healthcare). The dissociation constant, Kd, was calculated from the ratio of dissociation rate (koff) and association rate (kon).
Cell based assays
Neutralization assays for IGF-IR, VEGFR2 and downstream kinases
BxPC-3 or MCF-7 cells seeded at 3×105 cells/well with 75% confluence were pre-incubated in serum-free medium for 18 hours. 100 nM ID2 or control treatment mixed with 80 ng/ml IGF-I was incubated with the appropriate cells at 37 °C for 15 minutes. The cells were lysed in lysis buffer containing 50 mM Tris-Cl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1.25% CHAPS, 1× complete protease inhibitor cocktail (Roche #04693132001), and 1× PhosSTOP phosphatase inhibitor cocktail (Roche #04906837001), followed by a standard immunoblotting procedure. Phosphorylation of IGF-IR, ERK1/2 and AKT was detected with rabbit anti p-IGF-IR (cell signaling #3024), rabbit anti p-ERK1/2 (cell signaling #9101) and rabbit anti p-AKT (cell signaling #4060) antibodies, respectively. Total ERK1/2 and AKT were detected by mouse anti-ERK1/2 (cell signaling #4696) and mouse anti-pan AKT (cell signaling #2920) antibodies, respectively. Blots were stained with secondary antibodies IRDye 800CW goat anti rabbit IgG (Li-Cor #926–32211) and IRDye 680LT goat anti mouse IgG (Li-Cor #926–32220), and signals were detected by an Odyssey Fluorescence Imaging System. For total IGF-IR detection, the membrane was stripped and re-probed with rabbit anti IGF-IR Ab (cell signaling #3018) and IRDye 800CW goat anti rabbit IgG (Li-Cor #926–32211).
PAE cells overexpressing KDR (human VEGFR2) were plated at 85%–90% confluence and starved in serum-free medium overnight. The mixture of 100 nM ID2 or the appropriate control and 40 ng/mL human VEGF-165 (R&D 293-VE-010) was pre-incubated and applied to starved cells for 15 minutes at 37°C. Total cell extract was obtained using the Lysis Buffer described above. VEGFR2 was precipitated with an in-house anti-human VEGFR2 antibody followed by a standard immunoblotting procedure. Phospho-VEGFR2 was probed with mouse anti-phosphotyrosine, horseradish peroxidase (HRP)-conjugated antibody (Santa Cruz biotechnology sc-508-HRP). After the membrane was stripped, it was re-probed with a rabbit IgG directed against mouse VEGFR2 with cross-reactivity to human VEGFR2 (Santa Cruz Biotechnology sc-504) and followed by an HRP-conjugated goat anti-rabbit IgG (Santa Cruz Biotechnology sc-2054) to measure total VEGFR2. Phospho- and total AKT and ERK1/2 were measured as described in the previous section.
MCF-7 and HUVEC viability assays
MCF-7 cells were seeded in 96-well plates at 10,000 cells/well and cultured for 24 hours followed by overnight starvation. Serially diluted ID2, IR mAb and FcD2 from 300 nM mixed with 100 ng/ml IGF-I were added in duplicate wells and cultured for 2 d. The number of viable cells in each culture was quantified using a CellTiter-Glo luminescent assay kit following the manufacturer's instruction (Promega G7570). The chemiluminescent signal was detected using a luminometer (Perkin Elmer, Vector TM X5). The percentage of inhibition was calculated using the following formula:
Human umbilical vein endothelial cells (HUVEC, ATCC # PCS-100–010) were seeded at 3,000 cells/well in a 96-well collagen-coated plate (BD #35440) with 0.5% FBS + EBM-2 basal medium (Cell Application Inc. #22211), followed by incubation at 37°C with 5% CO2 overnight. The next day, serially diluted ID2 or an appropriate control was pre-incubated with 30 ng/ml of VEGF at room temperature for one hour. The mixture was added into plate containing the HUVEC and incubated for 72 hours, followed by detection using a CellTiter-Glo assay as described above. The percentage of cell viability was calculated as follows:
The measurements in duplicate for each viability assay were analyzed using GraphPad Prism 6 (GraphPad Software, Inc.).
IGF-IR and VEGF internalization and degradation assays
For IGF-IR internalization and degradation, 20 nM ID2 or appropriate control was mixed with 100 ng/ml VEGF and incubated with BxPC-3 at 37°C for 18 hours. IGF-IR from the cell lysate was measured following the procedure described for the IGF-IR and kinase neutralization assay. For β-actin detection, a mouse anti-β-actin antibody (Sigma A5441) and an IRDye 680LT goat anti-mouse IgG (Li-Cor #926–32220) were used.
For VEGF and IGF-IR internalization and degradation, A431/IGF-IR cells were seeded at 4 × 105 per well in 12-well plates to obtain 90% confluency. 200 μl of 10 nM ID2 or the appropriate control and 400 ng/ml VEGF were added to the cells and incubated at 37°C for 24 hours. Culture supernatant was collected and VEGF was detected by Western blotting using a rabbit anti-VEGF IgG (Santa Cruz Biotechnology sc-507) followed by a goat anti-rabbit IgG-HRP (Santa Cruz biotechnology sc-2054). IGF-IR and β-actin were quantified as described in the previous section.
PAE/KDR cell migration assay
PAE/KDR cells at a density of 25,000 cells/well were seeded in Oris Fibronectin coated 96-well plates (Platypus Technologies CMAFN5.101) and allowed to adhere. Stoppers were then removed from the migration zone. A mixture of 100 ng/ml VEGF with 100 nM ID2, IR mAb or FcD2 was added into each well. The plate was incubated at 37°C for 20 hours to allow cell migration. The cells were stained with 4 μg/ml Calcein AM fluorescent dye (BD 354216). The fluorescent signal was measured in each migration zone using a SpectraMax M5 microplate reader (Molecular Devices). Statistical analysis using student t test (GraphPad Prism 6) was performed against triplicate measurements.
ADSC and ECFC co-culture cord formation assay
Human ADSCs and ECFCs were purchased from Lonza (Allendale, NJ) and cultured as previously described. Citation36 ADSC and ECFC co-culture assays were performed in basal media (MCDB-131 medium with 30 µg/mL L-ascorbic acid 2-phosphate, 1 μM dexamethasone, 50 μg/mL tobramycin, 10 μg/mL r-transferrin AF, and 10 μg/mL insulin). ADSC cells were plated at 40,000 cells per well in 96-well plates and incubated overnight at 37°C, 5% CO2. The media was removed the next day and 4,000 ECFC cells were plated on the ADSC monolayer, incubated at 37°C, 5% CO2 for 3–6 hours to allow ECFC attachment, and VEGF at 20 ng/mL and inhibitors (2–5×) were added to achieve the indicated final concentrations. Co-cultures were grown for 3 d and were fixed, stained, and imaged as described.Citation36 Cord formation images were capture with a Cellomics Arrayscan VTI. Total tube area was calculated from 9 fields for each well with 3 wells for each treatment.
Fluorescence confocal microscopy studies
To measure trafficking and delivery of ID2 and VEGF to the lysosomal compartment, 1 mg/mL ID2 or 100 μg/mL VEGF was labeled with FITC (Life Technologies) in PBS. 100 nM ID2 – FITC, 1 μg/mL VEGF – FITC or a mixture of 100 nM ID2 and 1 μg/mL VEGF – FITC was added to a suspension of BxPC-3 cells with a density of 5 × 105 cells per well followed by the addition of 100 nM LysoTracker Red DND-99 (Life Technologies). The reagents and cell mixtures were incubated at 4°C or 37°C for 2 hours before being washed with PBS and fixed in 4% PFA. Fixed cell images were acquired with a Nikon-C1 confocal microscope and image analysis was performed using Nikon Elements according to the manufacturer's manual.
In vivo efficacy, dose frequency and mechanism of action studies
All experiments and procedures were approved by an Internal Animal Care and Use Committee and performed in accordance with the United States Department of Agriculture and the National Institute of Health policies regarding the humane care and use of laboratory animals.
For in vivo efficacy and dose frequency studies, Caki-1, Colo-205, HT-29, or MiaPaCa-2 cell suspensions were implanted subcutaneously (S.C.) at 5 × 106 cells in 50% Matrigel® Matrix (Corning, Tewksbury, MA) into female athymic (nu/nu) mice, aged 7–8 weeks, obtained from Charles River Laboratories (Wilmington, MA). When tumor volumes reached an average of 330–440 mm3 the animals were randomized by tumor volume into treatment and control groups. All dosing was administered intraperitoneally (I.P.) 3 times per week in efficacy studies or according to the indicated schedules in dose frequency studies. Tumor volumes and body weights were recorded twice weekly, using the formula (tumor length) × (tumor width)Citation2 × π/6 = tumor volume in mm3. Tumor inhibition % was calculated as 100 × (1 − ratio of the relative tumor volumes (RTV) in the experimental versus the control groups), with RTV = final mean tumor volume/initial mean tumor volume. Tests for effect of treatment on tumor growth were compared by Repeated Measures ANOVA using JMP Statistical software (v. 9.0.3; SAS Institute). Mice were housed under pathogen-free conditions in micro-isolator cages with laboratory chow and water available ad libitum.
For mechanism of action studies, Colo-205 cells were implanted S.C. into female athymic (nu/nu) mice as outlined above. When tumors reached an average volume of 390 mm3 they were randomized by tumor volume into 4 treatment groups: USP Saline control, IR mAb (35 mg/kg), FcD2 (15 mg/kg) and ID2 (35 mg/kg). The respective doses were based on molar equivalent weights. Dosing was administered I.P. on days 1, 3 and 5. For IGF-IR ELISA, lysates were made from the tumors excised on Days 2 and 7. ELISA against human IGF-IR was performed using the Total Insulin Panel kits (Meso Scale Discovery). For evaluation of human and mouse VEGF, plasma was collected at days 2 and 7 of treatment. Human and murine plasma VEGF was evaluated using Meso Scale Discovery kits (Meso Scale Discovery) according to the manufacturer's directions. For cleaved caspase-3 evaluation in tumors, the animals were sacrificed at day 2 or day 7 and the tumors were resected, fixed in formalin, and paraffin embedded. Cleaved caspase-3 staining was evaluated quantitatively using the Aperio XT ScanScope system (Leica Biosystems). Differences in percentage of cleaved caspase-3 positive cells between treatment groups were evaluated by One Way ANOVA followed by Fisher's LSD comparison in SigmaPlot (Systat Software). Treatment-related differences in human IGF-IR, human and murine VEGF were evaluated by 2-way ANOVA followed by Bonferroni post hoc tests in SigmaPlot (Systat Software).
PK study
For mouse PK, male CD-1 mice were administered with ID2 (30 mg/kg), IR mAb (30 mg/kg) or FcD2 (15 mg/kg) by intravenous bolus injection. Blood was collected over a 2 week period from 3 animals per group per time point and processed to serum. To characterize the serum PK of the treatment, a human IgG Fc ELISA was employed, with an affinity purified fragment of a goat anti-human IgG-Fc antibody (Bethyl Laboratories, Inc.) coated on the plate (Thermo Scientific™ Immulon® 4HBX) as the capture reagent. After incubation with serum samples, mouse anti-human IgG (Fc)-HRP (Southern Biotech) was employed as detection reagent. Immunoreactivity was determined from known amounts of ID2 in 20% mouse serum (Lampire Biological Laboratories) using a 5-parameter algorithm (StatLia, version 3.2).
Disclosure of Potential Conflicts of Interest
The authors are previous or current employees of ImClone Systems and/or Eli Lilly and Company.
Supplemental_Material.docx
Download MS Word (1.6 MB)Acknowledgments
We thank Xenia Luna for SPR support, Ana Cardoso for mammalian expression and Michelle Iacolina for sequencing service. We are grateful to Dan Lu, Dr. Jaafar Haidar, Dr. Kaori Hiraga and Dr. Gregory Plowman for insightful discussion.
Funding
The research and publication of this article are supported by funding from Lilly Research Laboratories.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Chames P, Baty D. Bispecific antibodies for cancer therapy: the light at the end of the tunnel? mAbs 2009; 1:539-47; PMID:20073127; http://dx.doi.org/10.4161/mabs.1.6.10015
- Kontermann RE, Brinkmann U. Bispecific antibodies. Drug Dis Today 2015; PMID:25728220
- Linke R, Klein A, Seimetz D. Catumaxomab: clinical development and future directions. mAbs 2010; 2:129-36; PMID:20190561; http://dx.doi.org/10.4161/mabs.2.2.11221
- Dong J, Demarest SJ, Sereno A, Tamraz S, Langley E, Doern A, Snipas T, Perron K, Joseph I, Glaser SM, et al. Combination of two insulin-like growth factor-I receptor inhibitory antibodies targeting distinct epitopes leads to an enhanced antitumor response. Mol Cancer Ther 2010; 9:2593-604; PMID:20716637; http://dx.doi.org/10.1158/1535-7163.MCT-09-1018
- Lu D, Zhang H, Ludwig D, Persaud A, Jimenez X, Burtrum D, Balderes P, Liu M, Bohlen P, Witte L, et al. Simultaneous blockade of both the epidermal growth factor receptor and the insulin-like growth factor receptor signaling pathways in cancer cells with a fully human recombinant bispecific antibody. J Biol Chem 2004; 279:2856-65; PMID:14576153; http://dx.doi.org/10.1074/jbc.M310132200
- Jordan JL, Arndt JW, Hanf K, Li G, Hall J, Demarest S, Huang F, Wu X, Miller B, Glaser S, et al. Structural understanding of stabilization patterns in engineered bispecific Ig-like antibody molecules. Proteins 2009; 77:832-41; PMID:19626705; http://dx.doi.org/10.1002/prot.22502
- Davis JH, Aperlo C, Li Y, Kurosawa E, Lan Y, Lo KM, Huston JS. SEEDbodies: fusion proteins based on strand-exchange engineered domain (SEED) CH3 heterodimers in an Fc analogue platform for asymmetric binders or immunofusions and bispecific antibodies. Protein Eng, Des Sel 2010; 23:195-202; http://dx.doi.org/10.1093/protein/gzp094
- Mabry R, Gilbertson DG, Frank A, Vu T, Ardourel D, Ostrander C, Stevens B, Julien S, Franke S, Meengs B, et al. A dual-targeting PDGFRbeta/VEGF-A molecule assembled from stable antibody fragments demonstrates anti-angiogenic activity in vitro and in vivo. mAbs 2010; 2:20-34; PMID:20065654; http://dx.doi.org/10.4161/mabs.2.1.10498
- Mabry R, Lewis KE, Moore M, McKernan PA, Bukowski TR, Bontadelli K, Brender T, Okada S, Lum K, West J, et al. Engineering of stable bispecific antibodies targeting IL-17A and IL-23. Protein Eng Des Sel 2010; 23:115-27; http://dx.doi.org/10.1093/protein/gzp073
- Miller BR, Demarest SJ, Lugovskoy A, Huang F, Wu X, Snyder WB, Croner LJ, Wang N, Amatucci A, Michaelson JS, et al. Stability engineering of scFvs for the development of bispecific and multivalent antibodies. Protein Eng Des Sel 2010; 23:549-57; http://dx.doi.org/10.1093/protein/gzq028
- Schaefer G, Haber L, Crocker LM, Shia S, Shao L, Dowbenko D, Totpal K, Wong A, Lee CV, Stawicki S, et al. A two-in-one antibody against HER3 and EGFR has superior inhibitory activity compared with monospecific antibodies. Cancer Cell 2011; 20:472-86; PMID:22014573; http://dx.doi.org/10.1016/j.ccr.2011.09.003
- Lewis SM, Wu X, Pustilnik A, Sereno A, Huang F, Rick HL, Guntas G, Leaver-Fay A, Smith EM, Ho C, et al. Generation of bispecific IgG antibodies by structure-based design of an orthogonal Fab interface. Nat Biotechnol 2014; 32:191-8; PMID:24463572; http://dx.doi.org/10.1038/nbt.2797
- Von Kreudenstein TS, Escobar-Carbrera E, Lario PI, D'Angelo I, Brault K, Kelly J, Durocher Y, Baardsnes J, Woods RJ, Xie MH, et al. Improving biophysical properties of a bispecific antibody scaffold to aid developability: quality by molecular design. mAbs 2013; 5:646-54; PMID:23924797; http://dx.doi.org/10.4161/mabs.25632
- Schaefer W, Regula JT, Bahner M, Schanzer J, Croasdale R, Durr H, Gassner C, Georges G, Kettenberger H, Imhof-Jung S, et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc Natl Acad Sci U S A 2011; 108:11187-92; PMID:21690412; http://dx.doi.org/10.1073/pnas.1019002108
- Labrijn AF, Meesters JI, de Goeij BE, van den Bremer ET, Neijssen J, van Kampen MD, Strumane K, Verploegen S, Kundu A, Gramer MJ, et al. Efficient generation of stable bispecific IgG1 by controlled Fab-arm exchange. Proc Natl Acad Sci U S A 2013; 110:5145-50; PMID:23479652; http://dx.doi.org/10.1073/pnas.1220145110
- List T, Neri D. Immunocytokines: a review of molecules in clinical development for cancer therapy. Clin Pharmacol 2013; 5:29-45; PMID:23990735
- Ferrara N. VEGF-A: a critical regulator of blood vessel growth. Eur Cytokine Netw 2009; 20:158-63; PMID:20167554
- Gerber HP, Hillan KJ, Ryan AM, Kowalski J, Keller GA, Rangell L, Wright BD, Radtke F, Aguet M, Ferrara N. VEGF is required for growth and survival in neonatal mice. Development 1999; 126:1149-59; PMID:10021335
- Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol 2005; 23:1011-27; PMID:15585754; http://dx.doi.org/10.1200/JCO.2005.06.081
- Senger DR, Van de Water L, Brown LF, Nagy JA, Yeo KT, Yeo TK, Berse B, Jackman RW, Dvorak AM, Dvorak HF. Vascular permeability factor (VPF, VEGF) in tumor biology. Cancer Metastasis Reviews 1993; 12:303-24; PMID:8281615; http://dx.doi.org/10.1007/BF00665960
- Claesson-Welsh L. Healing hemangiomas. Nat Med 2008; 14:1147-8; PMID:18989275; http://dx.doi.org/10.1038/nm1108-1147
- Wiesmann C, Fuh G, Christinger HW, Eigenbrot C, Wells JA, de Vos AM. Crystal structure at 1.7 A resolution of VEGF in complex with domain 2 of the Flt-1 receptor. Cell 1997; 91:695-704; PMID:9393862; http://dx.doi.org/10.1016/S0092-8674(00)80456-0
- Holash J, Davis S, Papadopoulos N, Croll SD, Ho L, Russell M, Boland P, Leidich R, Hylton D, Burova E, et al. VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc Natl Acad Sci U S A 2002; 99:11393-8; PMID:12177445; http://dx.doi.org/10.1073/pnas.172398299
- Burtrum D, Zhu Z, Lu D, Anderson DM, Prewett M, Pereira DS, Bassi R, Abdullah R, Hooper AT, Koo H, et al. A fully human monoclonal antibody to the insulin-like growth factor I receptor blocks ligand-dependent signaling and inhibits human tumor growth in vivo. Cancer Res 2003; 63:8912-21; PMID:14695208
- Rowinsky EK, Youssoufian H, Tonra JR, Solomon P, Burtrum D, Ludwig DL. IMC-A12, a human IgG1 monoclonal antibody to the insulin-like growth factor I receptor. Clin Cancer Res 2007; 13:5549s-55s; PMID:17875788; http://dx.doi.org/10.1158/1078-0432.CCR-07-1109
- Yap TA, Olmos D, Molife LR, de Bono JS. Targeting the insulin-like growth factor signaling pathway: figitumumab and other novel anticancer strategies. Exp Opin Investig Drugs 2011; 20:1293-304; PMID:21777167; http://dx.doi.org/10.1517/13543784.2011.602630
- Li H, Adachi Y, Yamamoto H, Min Y, Ohashi H, Ii M, Arimura Y, Endo T, Lee CT, Carbone DP, et al. Insulin-like growth factor-I receptor blockade reduces tumor angiogenesis and enhances the effects of bevacizumab for a human gastric cancer cell line, MKN45. Cancer 2011; 117:3135-47; PMID:21264842; http://dx.doi.org/10.1002/cncr.25893
- Reinmuth N, Liu W, Fan F, Jung YD, Ahmad SA, Stoeltzing O, Bucana CD, Radinsky R, Ellis LM. Blockade of insulin-like growth factor I receptor function inhibits growth and angiogenesis of colon cancer. Clin Cancer Res 2002; 8:3259-69; PMID:12374697
- Spiliotaki M, Markomanolaki H, Mela M, Mavroudis D, Georgoulias V, Agelaki S. Targeting the insulin-like growth factor I receptor inhibits proliferation and VEGF production of non-small cell lung cancer cells and enhances paclitaxel-mediated anti-tumor effect. Lung Cancer 2011; 73:158-65; PMID:21190751; http://dx.doi.org/10.1016/j.lungcan.2010.11.010
- Feldser D, Agani F, Iyer NV, Pak B, Ferreira G, Semenza GL. Reciprocal positive regulation of hypoxia-inducible factor 1alpha and insulin-like growth factor 2. Cancer Res 1999; 59:3915-8; PMID:10463582
- Kim KW, Bae SK, Lee OH, Bae MH, Lee MJ, Park BC. Insulin-like growth factor II induced by hypoxia may contribute to angiogenesis of human hepatocellular carcinoma. Cancer Res 1998; 58:348-51; PMID:9443416
- Harding TC, Long L, Palencia S, Zhang H, Sadra A, Hestir K, Patil N, Levin A, Hsu AW, Charych D, et al. Blockade of nonhormonal fibroblast growth factors by FP-1039 inhibits growth of multiple types of cancer. Sci Transl Med 2013; 5:178ra39; PMID:23536011; http://dx.doi.org/10.1126/scitranslmed.3005414
- Ferrara N, Chen H, Davis-Smyth T, Gerber HP, Nguyen TN, Peers D, Chisholm V, Hillan KJ, Schwall RH. Vascular endothelial growth factor is essential for corpus luteum angiogenesis. Nat Med 1998; 4:336-40; PMID:9500609; http://dx.doi.org/10.1038/nm0398-336
- Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med 1999; 5:623-8; PMID:10371499; http://dx.doi.org/10.1038/9467
- Sullivan LA, Brekken RA. The VEGF family in cancer and antibody-based strategies for their inhibition. mAbs 2010; 2:165-75; PMID:20190566; http://dx.doi.org/10.4161/mabs.2.2.11360
- Falcon BL, O'Clair B, McClure D, Evans GF, Stewart J, Swearingen ML, Chen Y, Allard K, Lee LN, Neote K, et al. Development and characterization of a high-throughput in vitro cord formation model insensitive to VEGF inhibition. J Hematol Oncol 2013; 6:31; PMID:23622716; http://dx.doi.org/10.1186/1756-8722-6-31
- Rudge JS, Holash J, Hylton D, Russell M, Jiang S, Leidich R, Papadopoulos N, Pyles EA, Torri A, Wiegand SJ, et al. VEGF Trap complex formation measures production rates of VEGF, providing a biomarker for predicting efficacious angiogenic blockade. Proc Natl Acad Sci U S A 2007; 104:18363-70; PMID:18000042; http://dx.doi.org/10.1073/pnas.0708865104
- Witte L, Hicklin DJ, Zhu Z, Pytowski B, Kotanides H, Rockwell P, Bohlen P. Monoclonal antibodies targeting the VEGF receptor-2 (Flk1/KDR) as an anti-angiogenic therapeutic strategy. Cancer Metastasis Rev 1998; 17:155-61; PMID:9770111; http://dx.doi.org/10.1023/A:1006094117427
- Shen Y, Zeng L, Zhu A, Blanc T, Patel D, Pennello A, Bari A, Ng S, Persaud K, Kang YK, et al. Removal of a C-terminal serine residue proximal to the inter-chain disulfide bond of a human IgG1 lambda light chain mediates enhanced antibody stability and antibody dependent cell-mediated cytotoxicity. mAbs 2013; 5:418-31; PMID:23567210; http://dx.doi.org/10.4161/mabs.24291
- Yang Y, Zhang Y, Cao Z, Ji H, Yang X, Iwamoto H, Wahlberg E, Lanne T, Sun B, Cao Y. Anti-VEGF- and anti-VEGF receptor-induced vascular alteration in mouse healthy tissues. Proc Natl Acad Sci U S A 2013; 110:12018-23; PMID:23818623; http://dx.doi.org/10.1073/pnas.1301331110
- Bogusz J, Majchrzak A, Medra A, Cebula-Obrzut B, Robak T, Smolewski P. Mechanisms of action of the anti-VEGF monoclonal antibody bevacizumab on chronic lymphocytic leukemia cells. Postepy Hig Med Dosw (Online) 2013; 67:107-18; PMID:23475487; http://dx.doi.org/10.5604/17322693.1038349
- Stefanini MO, Wu FT, Mac Gabhann F, Popel AS. Increase of plasma VEGF after intravenous administration of bevacizumab is predicted by a pharmacokinetic model. Cancer Res 2010; 70:9886-94; PMID:21118974; http://dx.doi.org/10.1158/0008-5472.CAN-10-1419
- Bottos A, Martini M, Di Nicolantonio F, Comunanza V, Maione F, Minassi A, Appendino G, Bussolino F, Bardelli A. Targeting oncogenic serine/threonine-protein kinase BRAF in cancer cells inhibits angiogenesis and abrogates hypoxia. Proc Natl Acad Sci U S A 2012; 109:E353-9; PMID:22203991; http://dx.doi.org/10.1073/pnas.1105026109
- Igawa T, Maeda A, Haraya K, Tachibana T, Iwayanagi Y, Mimoto F, Higuchi Y, Ishii S, Tamba S, Hironiwa N, et al. Engineered monoclonal antibody with novel antigen-sweeping activity in vivo. PloS one 2013; 8:e63236; PMID:23667591; http://dx.doi.org/10.1371/journal.pone.0063236
- Kang YK, Hamzik J, Felo M, Qi B, Lee J, Ng S, Liebisch G, Shanehsaz B, Singh N, Persaud K, et al. Development of a novel and efficient cell culture flocculation process using a stimulus responsive polymer to streamline antibody purification processes. Biotechnol Bioeng 2013; 110:2928-37; PMID:23740533; http://dx.doi.org/10.1002/bit.24969