
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.abstract
Antibody humanization describes the procedure of grafting a non-human antibody's complementarity-determining regions, i.e., the variable loop regions that mediate specific interactions with the antigen, onto a β-sheet framework that is representative of the human variable region germline repertoire, thus reducing the number of potentially antigenic epitopes that might trigger an anti-antibody response. The selection criterion for the so-called acceptor frameworks (one for the heavy and one for the light chain variable region) is traditionally based on sequence similarity. Here, we propose a novel approach that selects acceptor frameworks such that the relative orientation of the 2 variable domains in 3D space, and thereby the geometry of the antigen-binding site, is conserved throughout the process of humanization. The methodology relies on a machine learning-based predictor of antibody variable domain orientation that has recently been shown to improve the quality of antibody homology models. Using data from 3 humanization campaigns, we demonstrate that preselecting humanization variants based on the predicted difference in variable domain orientation with regard to the original antibody leads to subsets of variants with a significant improvement in binding affinity.
Abbreviations
| mAb | = | monoclonal antibody |
| CDR | = | complementarity-determining region |
| ECD | = | extracellular domain |
| HCV | = | hepatitis virus C |
| PDB | = | Protein Data Bank |
| LEL | = | large extracellular loop |
Introduction
Therapeutic monoclonal antibodies (mAbs)Citation1-3 of xenogeneic origin can trigger immune reactions when administered to human patients. Immune reactions directed against the mAb can lead to loss of efficacy of the drug and, even more important, to adverse effects ranging from mild local skin reactions to life-threatening acute anaphylaxis and systemic inflammatory response syndrome.Citation4 Based on the reasoning that mAbs that are typically human in sequence are less likely to trigger the human immune response, it has become common practice to replace large parts of the original non-human (typically rodent) antibody with counterparts naturally occurring in human, thus reducing the number of potentially antigenic epitopes. To increase the “humanness” of a given non-human antibody while preserving its original antigen binding properties, 2 major engineering strategies are known. The first strategy, chimerization, describes the procedure of grafting the variable regions of the non-human antibody onto the constant regions of a human antibody. During this process, the human IgG subtype and corresponding Fc effector functions can be chosen to best match the desired drug profile of the mAb. The resulting chimeric antibody loses the antigenicity conferred by the non-human constant regions, but retains significant xenogeneic content. By contrast, the second strategy, humanization, involves discarding all but the antigen-binding parts of the original antibody by grafting only the non-human complementarity-determining regions (CDRs) onto the conserved β-sheet framework of a human variable region (typically a homolog of the non-human donor variable region). In combination with the constant regions of a human IgG, only the antigenicity conferred by the CDRs remains. Both chimerization and humanization are enabled by the modular, robust and highly conserved structure that is a characteristic of antibodies. While it is known that the immunogenicity of mAbs is not a simple function of the degree of sequence identity to human antibodies, nor of antigenicity (e.g., the number of predicted T-cell epitopes), and that even so-called “fully human” antibodies derived from transgenic animals or phage display are able to elicit a human anti-antibody response, humanization is recognized as a standard procedure to at least manage this risk. An overview of which marketed mAbs are non-human, chimeric, humanized or fully human can be found in Ref. 3.
The initial step of humanization is the categorization of the antibody's variable region sequences into framework and CDR segments. The definition of these segments is typically adopted from the pioneering work of KabatCitation5 and Chothia.Citation6 Based on the degree of sequence homology to the non-human donor framework, a suitable human acceptor framework is identified, either from among the entirety of known human antibody sequences, or a set of human V-region germline sequences. The CDR sequence segments of the non-human antibody are then inserted into the human donor framework sequences. Finally, the humanized sequences can be refined by introducing either forward mutations (non-human CDR residues are substituted to match the human antibody repertoire) or backward mutations (human acceptor framework residues are substituted to match the non-human origin). For a comprehensive overview of different humanization methodologies, a number of reviews are available.Citation7-12
Exchanging the framework regions of an antibody in the process of humanization has a potential effect on how the CDRs are presented to the antigen, and thus on the antigen-binding properties. While murine and human variable regions typically show a high degree of homology, rabbit variable regions tend to exhibit species-dependent characteristics such as deletions in the β-sheet framework that can be difficult to emulate using the human V-region germline repertoire. Therefore, a low to moderate loss of binding affinity of the humanized antibody is often considered tolerable. To increase the probability of obtaining a satisfactory humanized version of the original antibody, it is common practice to generate multiple humanization variants of the heavy chain and light chain variable regions (VH and VL), differing, e.g., in the choice of acceptor framework, or the number and location of forward and backward mutations. Those variants are then expressed and evaluated in a matrix, i.e., considering all possible pairings between VH and VL, from which the best candidate can be picked based on its binding properties. Recent publications also highlight the value of crystal structures or accurate homology models to improve the “success rate” of antibody humanization.Citation13-15 If the non-human antibody-antigen complex structure is available, backward and forward mutations can be introduced much more precisely than on the mere sequence level. Homology models of the humanized antibody, in turn, can help to detect problematic amino acid substitutions. By now, a number of in silico tools is available to guide the humanization processCitation16,17 and to provide automatically generated antibody Fv models.Citation18
In the following, we focus on an aspect of antibody structure that, in the context of humanization, is often overlooked: the relative orientation of VH and VL domain. A survey of the known repertoire of antibody crystal structures reveals a notable variability in the parameters of VH-VL orientation,Citation19–21 and it seems likely that modulating VH-VL orientation not only is a necessary means to accommodate the diverse antigenic shapes that antibodies are confronted with, but also a mechanism to further diversify the composition (and thus increase the possible number) of antibody paratopes - in addition to the well-known mechanisms of diversification involving variations in length and sequence of the CDRs. It is reasonable to assume that changes in VH-VL orientation (e.g., caused by exchanging the β-sheet framework during humanization) might induce changes with regard to antigen binding.
Here, we present an in silico methodology that aims at preselecting humanization variants that are likely to preserve the VH-VL orientation of the reference antibody. For this purpose, we characterize VH-VL orientation in terms of the 6 ABangle orientation parameters derived by Dunbar et al.Citation21 (). To predict the ABangle parameters for a given non-human antibody and its humanization variants, we use a machine learning approach that was published recently.Citation22 The random forest model predictor evaluates a set of 54 conserved residues (the “orientation fingerprint”) at the interface of VH and VL to derive an estimate for each of the 6 ABangle parameters (). Once the putative ABangle parameters for the reference antibody and its humanization variants have been determined, each variant can be ranked with regard to the expected difference in VH-VL orientation. Finally, the humanization variants that are expected to exhibit a markedly different VH-VL orientation than the reference antibody can be discarded so that the overall number of variants to be cloned, expressed and tested is reduced.
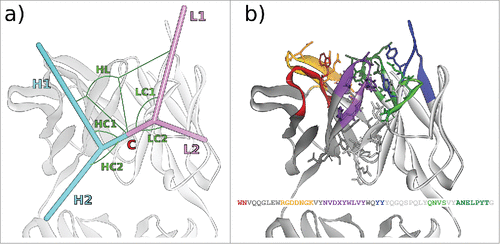
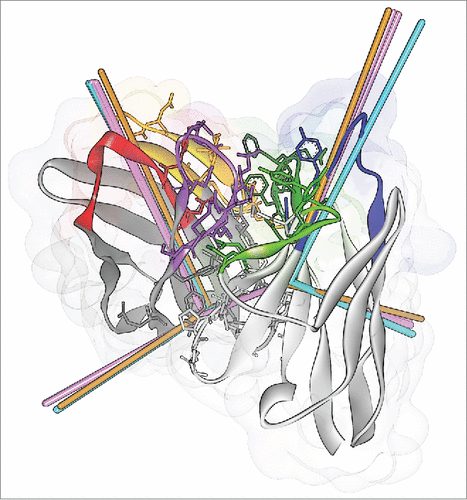
Figure 1. (A) The 6 ABangle VH-VL parameters that describe the variable degrees of freedom of VH-VL orientation consist of the torsion angle HL, from H1 to L1 measured about C, the bend angle HC1, between H1 and C, the bend angle HC2, between H2 and C, the bend angle LC1, between L1 and C, the bend angle LC2, between L2 and C, and the length of C, dc (not shown). (B) Residues belonging to the VH-VL orientation fingerprint, shown in stick representation, and the resulting fingerprint string. CDR color coding follows IMGT/Collier-de-PerlesCitation23 conventions: CDR-H1 is colored red, CDR-H2 orange, CDR-H3 purple, CDR-L1 blue, CDR-L2 light green, and CDR-L3 dark green. The example shows the crystal structure with PDB ID 3PP4.Citation24
To test our hypothesis that humanization variants that are likely to preserve the VH-VL orientation parameters of the original antibody are also likely to exhibit favorable antigen-binding properties, we retrospectively apply our method to 3 humanization campaigns for which experimentally derived binding affinities for all sequence variants (i.e., the complete matrix of all VH and VL combinations) are available. We apply different filtering algorithms that consider the predicted difference in VH-VL orientation with regard to the reference antibody, and evaluate the quality of the remaining subsets of humanization variants in terms of the average binding affinity.
The exemplary humanization campaigns presented here involve 2 murine antibodies directed against the extracellular domain (ECD) of CD81 receptor, and a rabbit antibody directed against phosphorylated tau protein (tau/pS422). Antibodies targeting CD81, a receptor required by hepatitis C virus (HCV) to infect human hepatocytes, were recently described by Vexler et al.Citation25 and Ji et al.Citation26 Two of these high affinity anti-CD81 mAbs, CD81K04 and CD81K13, showed potent and broad spectrum antiviral activity in various in vitro assays and were humanized. The anti-CD81 mAb CD81K04 could completely block HCV infection and spread in vivo, indicating that CD81 is essential for HCV-mediated pathology in the liver.Citation26 Tau, by contrast, is an axonal protein that normally associates with microtubules and thereby stabilizes them. In Alzheimer disease, abnormal phosphorylation, misfolding and aggregation of tau lead to neurofibrillary tangle formation, and ultimately to neuronal cell death.Citation27 A proposed mode of action of the anti-tau/pS422 antibody Rb86 is that it binds to membrane-associated tau/pS422, upon which the antibody-antigen complexes are internalized and cleared within the cell, so that the neurodegenerative pathology of tau is ameliorated. An alignment of the VH and VL sequences of CD81K04, CD81K13 and Rb86 is given in .
Figure 2. VH (top) and VL (bottom) sequences of the 3 rodent antibodies CD81K04, CD81K13, and Rb86. Framework and CDR classification follows WolfGuy nomenclature. Sequence positions that are part of the VH-VL orientation fingerprint are highlighted with a gray background. VH-VL orientation fingerprint positions that are unpopulated in a given antibody are denoted with the letter X. CDR color coding as described in Fig. 1.

To complement our humanization study and the analysis of VH-VL orientation, the crystal structures of the 3 rodent antibodies in complex with their respective antigen have been determined and deposited in the Protein Data Bank (PDB) (www.rcsb.org; PDB IDs 5DFV, 5DFW, 5DMG).
Results
Crystal structures
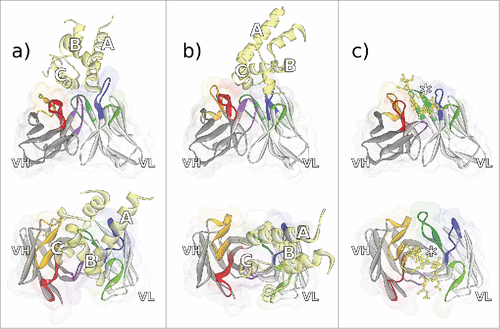
The crystal structures of the 2 murine antibodies CD81K04 and CD81K13 binding to the large extracellular loop (LEL) of the CD81 receptor ECD are depicted in . The crystal structure of CD81 LEL in the apo form had been determined previously.Citation28,29 Both anti-CD81 antibodies can be considered examples of mAbs that recognize a conformational epitope, i.e., an epitope that has a predefined, rigid, and often very complex shape. By contrast, the rabbit antibody Rb86 recognizes a peptide segment of tau protein (including the phosphorylation site pS422 that has been linked to tau pathology) that upon binding is still largely flexible in its conformation. The crystal structure of Rb86 binding to the peptide 15mer SID[MVDpSPQLATLAD] comprising residues 416–430 of tau/pS422 is shown in C. Only the residues shown in parentheses were assignable from the electron density.
Figure 3. Fv regions of the antibodies a) CD81K04 (PDB ID 5DFV), b) CD81K13 (PDB ID 5DFW), and c) Rb86 (5DMG) in complex with their respective antigen (colored yellow), shown in frontal (top) and top view (bottom). Capital letters in panels a) and b) indicate the helix names of CD81 LEL. In panel c), the location of phosphoserine in the peptide derived from tau/pS422 is highlighted with an asterisk. CDR color coding as described in Fig. 1.
In the process of antibody maturation, mainly amino acids in the CDRs are selected to recognize their antigen in a highly specific manner. In the case of CD81K04, CD81K13 and Rb86, the paratope, defined based on a 4 Å atom-atom distance cut-off to define antibody-antigen interactions, comprises amino acids from at least 5 of the 6 CDRs. Consequently, for these 3 antibodies, VH-VL orientation should be a potential codeterminant of antibody-antigen recognition, as each change in VH-VL orientation is prone to modulate the relative orientation of CDRs L1-L3 with regard to CDRs H1-H3, thus resculpting the shape of the paratope.
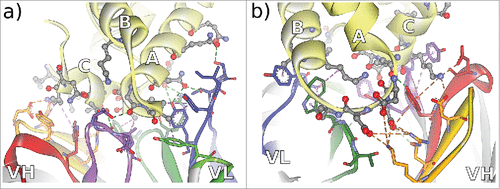
Using a spacious paratope formed by 18 residues in all of the 6 CDRs, CD81K04 targets a large conformational epitope formed by 16 residues of the CD81 receptor LEL, comprising a part of Helix A and almost the complete Helix C (). The epitope for CD81K13 is markedly different, with Helix B being recognized by the VL domain, and 2 loops, situated before Helix B and after Helix C, interacting with the VH domain (). Similarly to CD81K04, a relatively large number of 15 amino acids within the CDRs of CD81K13 interact with 13 residues of the CD81 LEL. CDR-L2 of CD81K13 is not in direct contact with the antigen, which sets it apart from the binding mode observable for CD81K04.
Figure 4. Non-bonded interactions at the antibody-CD81 LEL interface of antibodies a) CD81K04 and b) CD81K13. Capital letters indicate the helix names of CD81 LEL. Hydrogen bonds indicated as green dotted lines, hydrophobic interactions indicated as magenta dotted lines, and electrostatic interactions indicated as orange dotted lines. CDR color coding as described in Fig. 1.
Between two structured protein domains such as CD81 LEL and the respective anti-CD81 antibody, in general a broad contact surface can be established. The situation is different for antibodies binding smaller antigens, such as haptens and peptides, where in most cases, to maximize the number of possible antibody-antigen contacts, the binding site rather takes the shape of a deep groove. This implies that paratopic residues may not only originate from the CDRs, but also from the β-sheet framework of the antibody. This is also true for the peptide binding antibody Rb86 presented here.()
In the case of antibody Rb86, 4 framework residues (WolfGuy/Chothia: K332/H94, W401/H103, R612/L46, and Y615/L49) are in direct contact with tau peptide. Two more framework residues with bulky sidechains, W212/H47 and F602/L36, are instrumental for defining the topology of the paratope by limiting the size of the binding groove. Together with 18 residues situated in all of the 6 CDRs, they form a strong network to stabilize 10 consecutive amino acids of the peptide, including the phosphorylation site pS422. Only residue V420 of the peptide is not involved in immediate interactions with the paratope.
As a general rule of humanization, all paratopic residues of the reference antibody are to be retained, even if they are part of the β-sheet framework. In these cases, backward mutations in the human acceptor framework might be required. The prerequisite for taking such decisions is the availability of the antibody-antigen complex structure, or a very accurate model thereof. In silico tools such as Antibody i-PatchCitation30 can also help to identify residues that are likely to form a part of the paratope.
Each of the 3 original antibodies shows a distinct profile in terms of VH-VL orientation. A graphical representation of the ABangle vectors used to calculate the orientation measures (cp. ) is shown in . The absolute ABangle values for the 3 crystal structures can be found in Table SI1.
Figure 5. Non-bonded interactions at the interface of antibody Rb86 and the peptide derived from tau/pS422 from 2 different perspectives a) facing the VL interface, and b) facing the VH interface. From the co-crystallized 15-mer 416–430 of tau, only the segment 419–430 (MVDpSPQLATLAD) was assignable. Hydrogen bonds indicated as green dotted lines, hydrophobic interactions indicated as magenta dotted lines, and electrostatic interactions indicated as orange dotted lines. CDR color coding as described in Fig. 1.
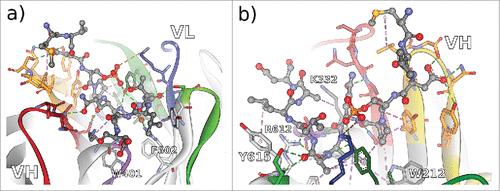
Figure 6. ABangle VH-VL orientation vectors calculated from the crystal structures of CD81K04 (cyan), CD81K13 (copper) and Rb86 (pink). The ribbon representation shows the crystal structure of CD81K04. Residues included in the VH-VL orientation fingerprint are shown in stick representation. Multiple vectors of the same color refer to multiple copies of the same Fv in the asymmetric unit of the respective crystal structure. CDR color coding as described in Fig. 1.
The cumulative distance in ABangle VH-VL orientation space (distABangle) between CD81K04 and CD81K13 is 15.91, and 7.94 between CD81K04 and Rb86. With a distABangle of 16.26, CD81K13 is also markedly different from Rb86. For comparison, the average distABangle between the multiple sequence-identical copies of CD81K04 and Rb86 within the asymmetrical unit of the crystal structure is 1.58 and 2.28, respectively, and the largest distABangle between any 2 antibody crystal structures that we are currently aware of is 37.66 (found for the crystal structures with PDB IDs 3MNV_BA and 4M1D_IM; trailing capital letters indicate the PDB chain identifiers associated with heavy and light chain). The rather unique VH-VL orientation of antibody CD81K13 becomes more apparent by comparing it against the background distribution of the redundant set of known antibody crystal structures, as is shown in .
Figure 7. The antibody crystal structures of CD81K04 (cyan), CD81K13 (copper) and Rb86 (pink) in ABangle VH-VL orientation space. For each of the 6 ABangle parameters, the background distribution of the redundant set of known antibody crystal structures is shown as a histogram. The predicted ABangle values are indicated by circles (leave-one-out predictor) and squares (all-knowing predictor).
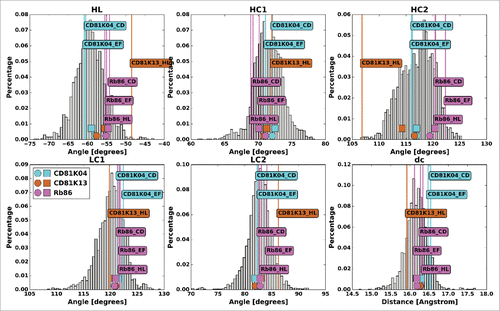
The murine antibody CD81K04 exhibits an average VH-VL orientation shared by the majority of known antibody crystal structures. Rb86, by contrast, shows comparatively high values for the torsion angle HL and the bend angle HC2, as well as a slightly smaller than average bend angle HC1. CD81K13 is a clearly exceptional antibody in terms of VH-VL orientation, with extreme values for HL and HC2, and a rather atypical value of LC2. The only other known antibody with a similar VH-VL orientation as CD81K13 is the hapten-binder 19G2 (crystal structures with PDB IDs 1FL3, 1UB5, 1UB6, and 3CFB), but there is almost no similarity in the orientation fingerprint of the 2 (data not shown).
The residues that are influential for the different ABangle parameters have been discussed at length in Refs. 21 and 22. While the orientation fingerprint of CD81K13 is largely unremarkable, the positions 398/H101 and 733/L87, both listed among the top determinants for the ABangle parameters HL and HC2, are histidine residues, making this antibody unique among the known repertoire of antibody structures and offering a possible explanation for the rather atypical VH-VL orientation. While CD81K04 and CD81K13 bind to the CD81 LEL with practically the same affinity (KD 0.5 nM at 25°CCitation26), CD81K13 has a shorter CDR-H3 loop and lacks the long CDR-L1 that is critical for the interaction between CD81K04 and its epitope (cp. ). One might speculate that, during maturation, CD81K13 compensated a possible lack of plasticity given by the rather short CDRs by adapting a very particular VH-VL orientation. The VH-VL orientation of CD81K13 leads to the formation of a comparatively deep binding groove between the 2 variable domains that offers a perfect fit for Helix C and the loop turn between Helix A and B of the CD81 LEL (cp. ).
In addition to the measured ABangle values, shows, for each antibody, the predicted ABangle values derived from the VH-VL orientation fingerprint, once for the predictor learned without the crystal structures of CD81K04, CD81K13 and Rb86 (subsequently referred to as “leave-one-out predictor," emulating the more common scenario where the crystal structure of the antibody to be humanized is not available), and once for a predictor that is aware of the VH-VL orientation parameters associated with the respective orientation fingerprints (subsequently referred to as “all-knowing” predictor).
For the leave-one-out predictor, the mean error of the predicted values is 6.53, mainly caused by a largely incorrect prediction of the ABangle values of CD81K13, with a cumulative error of 14.17, as opposed to 2.87 for CD81K04, and 2.55 for Rb86. Not surprisingly, the prediction error can be reduced significantly when the orientation fingerprint of the structures in question is included in the training set: The all-knowing predictor learned with the crystal structures of CD81K04, CD81K13 and Rb86 predicts the ABangle values with a cumulative error of 2.19, 10.82, and 1.24, respectively, leading to a mean error of 4.75. The prediction errors were determined in terms of distABangle between measured and predicted ABangle values. The predicted values can also be found in Table S1.
Humanization candidate sequences
shows the humanized VH and VL sequence variants of CD81K04 that were produced using the general CDR grafting principle. The parental murine sequences are given for reference. At the time of humanization, none of the crystal structures had been solved. Therefore, the decision to apply backward or forward mutations was then based on a homology model of the Fv region, without information on the exact location, size and shape of the paratope. However, a precise homology model of the Fv region in combination with knowledge of the antigen type, or even location and shape of the epitope, can be of help to narrow down positions where amino acid substitutions might be critical. For the humanization of CD81K04, 12 variants of VH and 9 variants of VL were generated. While each of the VH and VL variants is unique in sequence, some of the variants share exactly the same orientation fingerprint (cp. ) and thus, putatively, will adopt the same VH-VL orientation parameters. In , residues that are part of the orientation fingerprint and have been changed with regard to the murine reference sequence are highlighted.
Figure 8. VH (top) and VL (bottom) sequences of CD81K04 and humanized sequence variants thereof. Framework and CDR classification follows WolfGuy nomenclature. CDR color coding follows IMGT/Collier-de-PerlesCitation23 conventions: CDR-H1 is colored red, CDR-H2 orange, CDR-H3 purple, CDR-L1 blue, CDR-L2 light green, and CDR-L3 dark green. Sequence positions that are part of the VH-VL orientation fingerprint are highlighted with a gray background. Sequence positions that are part of the VH-VL orientation fingerprint but deviate from the parental sequence are highlighted with a green background.

The humanization variants of CD81K13 and Rb86 were generated following the same principle and can be found in Fig. S1 and Fig S2 of the Supplemental Information, respectively.
Binding signal matrix and ABangle distance matrix
After the humanized sequence variants have been devised, they are translated into coding DNA and cloned into an expression vector that encodes the complete antibody heavy or light chain, including the corresponding human constant regions. For reference, the variable regions of the original non-human antibody are also included. The antibodies are then expressed in a matrix, combining all heavy chain with all light chain plasmids.
shows the binding cell enzyme-linked immunosorbent assay (ELISA) results for the humanization matrix of antibody CD81K04. The top left cell corresponds to the chimerized form of the murine antibody, i.e., the variable region is 100% murine and thus should be unchanged in its antigen-binding properties, while the constant part is 100% human. The remainder of the top row and the leftmost column correspond to half-humanized antibodies with either the heavy chain or the light chain being a murine-human chimera, whereas all other cells are linked to antibodies that have been fully humanized.
Table 1. Matrix of binding cell ELISA results for antibody CD81K04 (denoted as Ref.) and its humanization variants (unitless absorbance).
As can be expected, the highest binding signal is detected for the chimeric CD81K04 reference antibody listed in the top left cell of . While some of the half-humanized and fully-humanized antibodies achieve binding signals that are very close to the murine reference (e.g., huVH09-huVL03), one can also identify humanization variants that bind only poorly to CD81 LEL, either in all of the possible combinations (huVL06, huVL08, huVL09, and huVH06), or when combined with certain other humanization variants from the matrix (huVL04, huVL05, huVL07, and huVH05). For some of the fully-humanized antibodies, hardly any binding signal is detectable.
Following the same approach of arranging the humanization variants of VH and VL in a matrix, for each VH-VL pair it is possible to: 1) generate the orientation fingerprint, 2) predict the putative VH-VL orientation parameters, and 3) calculate distABangle with regard to the reference antibody. The latter value is then added to the corresponding cell of the matrix. The methodology requires only the sequence of the reference antibody and its variants because all ABangle values involved are predicted in silico (including the reference VH-VL orientation). shows the distABangle matrix for the humanization variants of CD81K04.
Table 2. distABangle matrix for the humanization variants of antibody CD81K04 (denoted as Ref.), calculated using the leave-one-out predictor.
Analogously to the binding cell ELISA matrix (), the top left cell of corresponds to the murine reference antibody. The remainder of the top row and the leftmost column correspond to half-humanized antibodies where only one variable domain has been humanized, whereas all other cells of the distABangle matrix are associated with fully-humanized antibodies. Depending on the types of residues that form the respective orientation fingerprint, certain combinations of VH and VL humanization variants generate large distABangle values, while others remain closer to the predicted reference values. The predicted distABangle values are ranging from 0.28 to 1.32 for the half-humanized VH-VL pairs, and from 0.62 to 1.59 for the fully-humanized VH-VL pairs. None of the variants is predicted to have exactly the same VH-VL orientation as the murine origin, which is consistent with the observation that each variant has a minimum of 2 amino acid substitutions at the domain interface (cp. ). With a maximum of only 1.59, the predicted absolute distABangle values are low, and in the range of what is found for sequence-identical copies of the same antibody within a given crystal structure. We assume that: 1) the machine learning-based predictor might understate the absolute ABangle values, as it tends to lean toward the mean of the distribution of the structures that it has been trained with, and 2) the factual change in VH-VL orientation might indeed be relatively subtle, as many of the more influential orientation fingerprint positions are part of the CDRs, and thus remain unchanged in the course of humanization.
The distABangle values shown in were computed using the leave-one-out predictor. To evaluate how far the predictor quality affects the results, we recalculated the matrix using the all-knowing predictor that was demonstrated to predict ABangle values that are closer to those of the CD81K04 crystal structure (cp. and ). The results are shown in .
Table 3. distABangle matrix for the humanization variants of antibody CD81K04 (denoted as Ref.), calculated using the all-knowing predictor.
The absolute distABangle values change quite visibly, and now range from 0.49 to 2.20 for the half-humanized VH-VL pairs, and from 0.82 to 2.84 for the fully-humanized VH-VL pairs. This indicates that the all-knowing predictor is more capable of stratifying the different variants in terms of the extent of change in VH-VL orientation to be expected, although it is not possible to verify this when the humanization variants’ crystal structures are not available. Despite the change in the absolute range of values, the general trend found for the different variants of VH and VL seems to be in agreement with the distABangle matrix calculated with the less accurate leave-one-out predictor.
Analogous pairs of binding signal and distABangle matrices for the antibodies CD81K13 and Rb86 can be found in Tables S2-S4 (CD81K04) and Tables S5-S8 (Rb86) of the Supplemental Information. For Rb86, the binding signal data consists of 2 matrices of binding late (BL) and half-life (t½) values derived from surface plasmon resonance (SPR) measurements. In the case of CD81K13, the binding signal data is derived from binding cell ELISA experiments equivalent to those conducted for antibody CD81K04.
Matrix relatedness and correlation assessment
To assess if it might be meaningful to preselect humanization variants based on the putative difference in VH-VL orientation with regard to the given reference antibody, we investigated if, retrospectively, a relatedness between the distABangle matrices and the available binding signal matrices for the 3 humanization campaigns can be established.
There are several different measures to quantify the degree of relatedness between 2 matrices. Often, this relatedness is termed correlation, which, in this context, is technically incorrect. All of the existing measures make certain assumptions on the shape and rank of the matrices, and the kind of data they are trying to describe. One example is the Mantel correlation,Citation31 which measures the relatedness between 2 distance matrices. While the distABangle matrix qualifies as a proper distance matrix, it is questionable if a matrix of binding signals from an ELISA experiment could be interpreted as distance matrix in the sense of the Mantel correlation.
Other measures for quantifying matrix relatedness try to correlate multivariate data sets. Examples of these are the RV coefficient,Citation32 and the correlation coefficient from the PROTEST method.Citation33 These methods evaluate the correlation between 2 data sets where, for each sample, multiple measurements are available, and therefore can be thought of as extensions of the standard univariate correlation coefficient. Again, our data sets do not necessarily fit into this description, and there is no compelling reason why one should define either the VH or VL humanization variants as samples. However, if one would aim at rejecting complete VH or VL variants rather that rejecting only certain VH-VL combinations (thereby advantageously reducing the number of antibody chains that need to be cloned and expressed), one might investigate exactly this, and consider each humanization variant of either VH or VL as an inseparable “sample," and the predicted distABangle values obtained for all of the possible pairings as the “measurements." Consequently, one would expect that a multivariate correlation between the distABangle values and the observed binding signals can be established.
states the RV coefficients and their associated p-values for the different pairs of matrices, where the RV coefficient is either calculated from the perspective of the VH variant as sample while perceiving the paired VL variants as multivariate measurements, or vice versa. The p-values are calculated via a permutation test, and indicate the probability of randomly reaching an RV coefficient as high as or higher than the one that has been calculated. For antibody Rb86, 2 separate data sets, one for the BL matrix and one for the t½ matrix, are tested.
Table 4. RV coefficients and associated p-values for the different binding signal matrix-distABangle matrix pairs.
For the VH humanization variants, a low but significant correlation can be found for CD81K13 and Rb86 (BL matrix). The same holds for the VL humanization variants of CD81K04. Using the all-knowing predictor, and thus more accurately predicted distABangle matrices, the correlation can be improved in 7 of the 8 cases (with the VL humanization variants of CD81K04 being the exception), albeit not to a very high level. In the case of antibody Rb86, where 2 separate matrices of binding signals are available, the t½ matrix correlation is slightly better for the VL variants, and notably worse for the VH variants.
A less restricted view on the data set would be to view each possible VH-VL combination as an individual. In that case, it makes sense to vectorize both matrices and calculate the Pearson correlation coefficient. Given that VH-VL orientation is largely governed by the complex interplay of a number of key residues at both sides of the VH-VL domain interface, the pair-based approach might be more meaningful than the sample-based approach that focuses on only one of the 2 domains at a time.
shows the Pearson correlation coefficient and the according p-value for the different data sets. The p-value indicates the probability to reach the calculated correlation under the null hypothesis of having no correlation between distABangle and binding signal.
Table 5. Pearson correlation coefficients and associated p-values for the different binding signal matrix-distABangle matrix pairs. The correlation is calculated on vectorised versions of the respective matrices.
All data sets show a low to moderate but significant correlation, which, by using the more accurate all-knowing ABangle predictor can be improved further. The exception is antibody CD81K04, where the Pearson correlation coefficient of -0.417 remains in the same order as the one obtained with the leave-one-out predictor (-0.440), possibly because there is only a very minor improvement regarding the quality of the ABangle prediction when the all-knowing predictor is being used (cp. Table SI1). Given the evidence for correlation between the individual entries of the vectorized matrices, it can be expected that methods which reject certain VH-VL combinations based on unfavorable distABangle predictions should have a positive effect on the quality of the remaining set of humanized antibodies.
Humanization candidate subset selection
In the following, we evaluate 3 intuitive methods for optimizing a given set of humanized antibodies with regard to its antigen-binding properties by identifying and discarding candidates that are likely to be poor preservers of the non-human antibody's original VH-VL orientation. These methods are: 1) rejecting a fixed percentage of candidates that represent the upper end of the distABangle spectrum, 2) rejecting VH and VL variants that have a high average distABangle value over all possible pairings, and 3), as a variant of the former method, rejecting combinations of VH and VL variants that have a high average distABangle value over all possible pairings. For simplicity, we denote the 3 approaches as “reject fixed percentage," “reject worst chains," and “reject worst chain combinations." Of course, many more approaches of subset rejection are conceivable.
For the 2 methods “reject bad chains” and “reject bad chain combinations” that use the average distABangle value over all possible pairings to decide whether a VH or VL variant should be kept or rejected, it is necessary to define a rejection threshold value. Due to the fact that the absolute distABangle range and distribution is highly dependent on the individual set of candidates, it is non-trivial to define a general rule for determining an optimal threshold value. This is illustrated in.
Figure 9. VH (top row) and VL (bottom row) humanization variants of CD81K04, CD81K13 and Rb86, sorted by average distABangle value over all possible pairings (all-knowing ABangle predictor). The letter R denotes the non-human reference VH or VL. The dashed line indicates 80% of the maximum distABangle value, the threshold used for automatic rejection of a given variant.
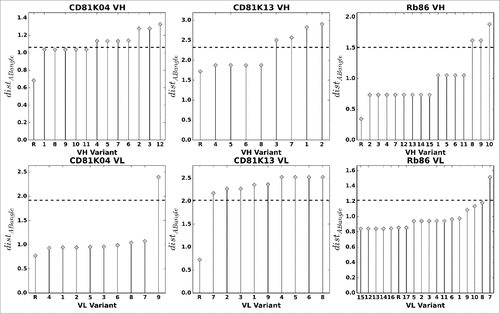
In some cases, a distinct stratification of different average distABangle levels is perceivable (e.g., Rb86 VH), so that a threshold value for variant rejection can be defined in a straightforward manner, while in other cases, the distribution of average distABangle values is almost continuous and subject to relatively minor variations (e.g., CD81K13 VL). Furthermore, in the case of antibody CD81K13, all humanization variants of VL are predicted to suffer from a notable change of VH-VL orientation, so that in principle one would either have to accept all variants (and risk possible negative effects on binding), or accept none (and define new ones that better conserve the VH-VL orientation fingerprint). Given the multitude of possible scenarios, defining the rejection threshold manually and on a case-by-case basis is recommended. Here, for the sake of comparability, we proceed with a fixed variant rejection threshold of 80% of the maximum average distABangle that is the same for our 3 example antibodies (cp. ).
illustrates the difference between the 3 subset rejection methods in the case of antibody CD81K04. Analogous representations for the antibodies CD81K13 and Rb86 can be found in Figs. S3, S4, and S5 of the Supplemental Information.
Figure 10. Overlay of the CD81K04 binding cell ELISA matrix (green indicates high binding signals, red low binding signals) with the selection matrix for each of the 3 rejection methods (darkened cells indicate rejected VH-VL pairs). The letter R denotes the murine reference VH or VL. Panel a) shows selections based on the leave-one-out predictor, panel b) shows selections based on the all-knowing predictor.
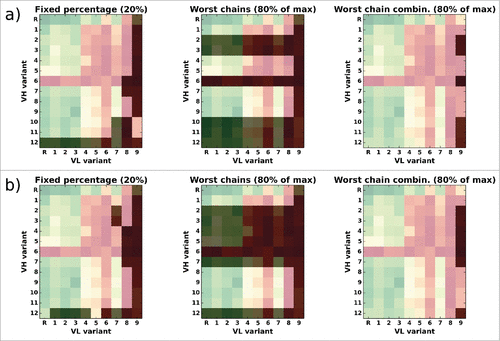
In this example, the method “reject worst percentage” (, left panel) rejects a fixed number of 26 VH-VL pairs from CD81K04's 13x10 candidate matrix, corresponding to the top 20% in terms of distABangle with regard to the reference. Of course, depending on the desired target number of candidates, a different percentage for rejection can be chosen. The selection varies slightly dependent on which version of the ABangle predictor is being used, but the rejected candidates mainly involve VH variant huVH12 and VL variant huVL09. The method “reject worst chains” discards one VL variant (huVL09) and a relatively high number of 6 (or 7, respectively) VH variants, so that only 63 (or 54, respectively) of the 130 possible VH-VL combinations remain. The method “reject worst chain combinations” is the least aggressive of the 3 approaches, and discards only 6 (or 7, respectively) of the 130 candidates, all of which involve VL variant huVL09.
To rate the performance of our different subset rejection methods, we calculate the ratio of the median binding signal between the kept and the rejected candidate subset, and, related to this ratio, a p-value based on a random permutation test to assess whether there would have been better possibilities to filter the set of humanization candidates. The results are shown in , respectively.
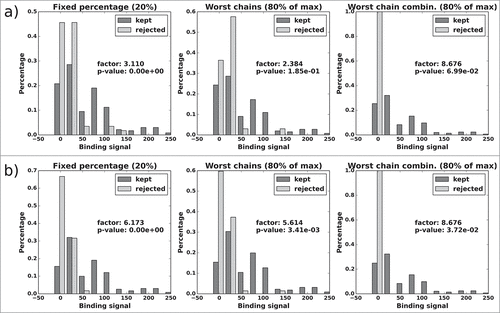
In the case of antibody CD81K04 (), all 3 selection methods deliver a kept candidate subset that has a 2.5 to 4.6-fold higher median binding signal than the rejected subset (see value “factor” in ). This is also evident from the histograms of the rejected candidate subsets, which tend to peak in the lower (left) region of the binding signal spectrum. None of the rejected subsets contains candidates from the highest range of binding signals (i.e., the rightmost region of the histogram). The results are highly significant for the “reject fixed percentage” selection method, and of low to moderate significance for the 2 methods that are dependent on averages over all possible pairings. This is coherent with our correlation analysis in the previous section, which showed better results for individual-based correlation than for sample-based correlation (i.e., considering whole rows and columns of the respective matrices at a time). Irrespective of the subset selection method, the binding signal factors and the p-values improve when the more accurate all-knowing ABangle predictor is being used.
Figure 11. CD81K04: Normalized histograms of the binding signal range of kept (dark gray) and rejected subset (light gray) for each of the 3 distABangle-based rejection methods. Binding signal values are taken from the CD81K04 binding cell ELISA matrix. Results in panel a) are based on selections made using the distABangle matrix from the leave-one-out predictor, while results in panel b) are based on selections made using the distABangle matrix from the all-knowing predictor.
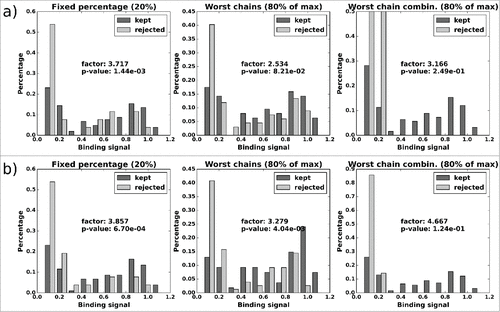
For CD81K13 (), the general picture is similar, but the improvement by candidate filtering is only very modest (1.6 to 1.8-fold). In this case, all 3 selection methods produce significant results, although the selection by “reject worst chains” is practically meaningless, as all VL variants but the reference are discarded when the fixed threshold for automatic rejection (80% of the maximum average distABangle value) is applied (cp. ). The set of rejected candidates then comprises up to 85 of the 90 VH-VL pairs, including some that are in the highest binding signal range. This underlines that it seems advisable to decide on a rejection threshold on a case-by-case basis and only after the individual distABangle distribution has been reviewed.
Figure 12. CD81K13: Normalized histograms of the binding signal range of kept (dark gray) and rejected subset (light gray) for each of the 3 distABangle-based rejection methods. Binding signal values are taken from the CD81K13 binding cell ELISA matrix. Panels a) and b) as described in Fig. 11.
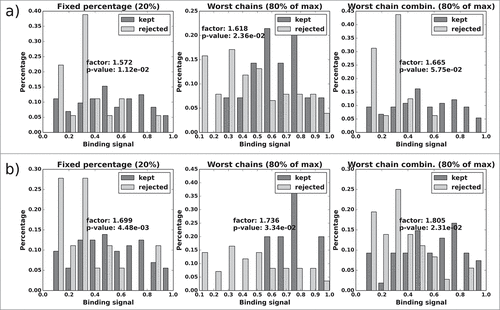
shows the results for antibody Rb86 using the BL matrix of binding signals. The three selection methods produce filtered sets of candidates with a 2.4 to 8.7-fold improvement in median binding signal, and candidates from the highest range of binding signals are never among the rejected VH-VL pairs. While the p-values for the “reject fixed percentage” method are flawless, the results for “reject worst chains” and “reject worst chain combinations” are not in the significant range, unless the all-knowing predictor is being used. As in the previous examples, the results (both binding signal ratio and p-value) improve visibly when the ABangle values are predicted more accurately (which we assume is the case for the all-knowing ABangle predictor).
Figure 13. Rb86: Normalized histograms of the binding signal range of kept (dark gray) and rejected subset (light gray) for each of the 3 distABangle-based rejection methods. Binding signal values are taken from the RB6 BL matrix. Panels a) and b) as described in .
An analogous representation for the half-life (t½) matrix of Rb86 can be found in Fig. S6 of the Supplemental Information. Note that for the t½ matrix, most binding signal ratios could not be determined, as the median binding signal of the rejected subset can come out zero. The Rb86 t½ matrix contains 84 entries that equal zero, which does not agree well with our method of analysis. As the subset selection is dependent only on the distABangle matrix, it is the same for both Rb86 binding signal matrices.
summarizes the results for the 3 humanization candidate rejection methods.
Table 6. Binding signal median change factor, p-value and number of total and rejected VH-VL pairs (“tot./rej.”) for each of the 3 humanization candidate selection methods.
Discussion
In all 3 examples, the method “reject fixed percentage” (20%) delivers a significant improvement in median binding signal of the remaining set of VH-VL pairs, which supports our hypothesis that selecting antibody humanization variants that are likely to preserve the VH-VL orientation of the original non-human antibody is meaningful. Even for antibody CD81K13, where the accuracy of the ABangle predictions is questionable, a moderate and significant improvement can be achieved. The quality of the results can be improved further when the more accurate predictor is used.
The method “reject worst chains” delivers similar improvement factors as the prior method, but achieves significant results only when combined with the all-knowing predictor. Thus, it seems to be more susceptible to inaccurate ABangle predictions. The main advantage of this method is that VH or VL variants are rejected as a whole, which is the optimal scenario in terms of potential for reducing wet lab workload. However, the absolute distABangle range and distribution is highly dependent on the individual set of VH-VL pairs, so that choosing the distABangle threshold value above which VH or VL variants should be rejected is not always straightforward. For example, using a rejection threshold corresponding to 80% of the maximum average distABanglevalue led to an excessive rejection of candidates in the case of antibody CD81K13, while it worked reasonably well for the other 2 examples.
The third method “reject worst chain combinations” is also dependent on a definite distABangle threshold value, but tries to minimize the risk of discarding good binders by rejecting only candidates were both VH and VL variant are found to be worse than the aforementioned threshold value. In practice, this leads to a very small number of rejected VH-VL pairs that, in 4 of the 6 evaluated cases, is not significantly better than a randomly selected subset of the same size. Furthermore, the method does not work in situations where one would wish to retain all variants of either VH or VL, and discard only variants of the complementary type. Finally, it is obvious that rejecting very small numbers of candidates is only of limited benefit when one aims at accelerating wet lab workflows in a noticeable manner.
Establishing a direct correlation between raw binding data and the predicted change in the VH-VL orientation (i.e., distABangle with regard to the non-human antibody) is obviously a challenging task. Firstly, there is a non-negligible intrinsic variation in the binding assays that are performed on supernatants or on micro-purified samples, and separating the effect of changes in VH-VL orientation from other factors that might have an effect on the binding signal is not altogether possible. Secondly, the predicted ABangle values may be subject to a significant error, in particular when the antibody to be humanized exhibits a very atypical VH-VL orientation (as is the case for the murine antibody CD81K13 presented here). Even for sequence-identical antibodies of known structure, notable deviations in the measured ABangle parameters have been found, which suggests that VH-VL orientation has an intrinsic variability and thus an unpredictable component (e.g., VH-VL orientation changes induced by subtle induced fit-like antigen-dependent effects) that cannot be tackled by a sequence-based machine learning approach.
Despite the fact that both binding signals as well as ABangle predictions can be significantly perturbed, our results show that a correlation between high distABangle values and low binding signals can be established. As our method is not dependent on absolute ABangle values but rather on differences in VH-VL orientation, we are confident that a part of the error cancels out, so that at least a qualitative categorization of the humanized antibodies into VH-VL orientation conservers and non-conservers is possible.
Nonetheless, and despite the fact that the humanized antibodies are ranked based on ABangle differences and not on absolute values, we found that using the more accurate ABangle predictions also led to better candidate selections, which was reflected by higher median binding signal factors and lower p-values. Therefore, the ABangle prediction accuracy remains an issue, and continuously retraining the ABangle predictor as new antibody crystal structures become available is mandatory. In the same line of thought, one might refrain from using the method in cases where the orientation fingerprint of the antibody to be humanized suggests that the prediction, due to a lack of known structures with a similar VH-VL interface, may be very inaccurate.
We have applied our analysis to finalized sets of humanization variants that were generated by the general CDR grafting procedure. The method can also be used to screen engineered antibody sequence variants in general for possible issues regarding the conservation of VH-VL orientation to avoid undesired effects on the antigen-binding properties or VH-VL stability. Finally, one could also envision beginning a humanization campaign for a given antibody by using the VH-VL orientation prediction method to find a favorable pair of human acceptor frameworks. An example for how this might work is illustrated in Figs. S7 and S8 of the Supplemental Information, where the CDRs of antibody CD81K04 were grafted in silico on a number of known VH and VL germline sequences, followed by a calculation of the resulting distABangle values with regard to the murine origin.
In contrast to the general CDR grafting routine, where, in principle, the sequences of VH and VL can be treated separately, this methodology always considers both variable domains simultaneously. In spirit, this idea is related to earlier attempts to identify preferences in VH-VL germline pairing;Citation34,35 however, the ABangle predictor has the advantage of being able to factor in the interplay between the residues of the conserved acceptor framework and the CDRs residues of the individual antibody to rate if VH-VL orientation will be preserved. This approach leads to humanized antibodies that, despite possibly having a lower sequence identity with the non-human origin, are more successful in preserving the original antigen-binding properties. Due to the fact that the method does not have to rely on backward mutations in the acceptor framework to recover the correct VH-VL orientation, the overall degree of humanness is likely to be improved.
Material and methods
Crystal structures
Co-crystallization of CD81K04 Fab fragment and CD81K31 (scFv) in complex with CD81 LEL
CD81LEL protein at 7.7 mg/ml concentration in 50 mM TRIS-Cl (pH 8.0), 300 mM sodium chloride was incubated on ice at 1:1 molar ratio with CD81K04 Fab fragment in 20 mM His, 140 mM NaCl (pH 6.0). Crystals of the CD81LEL-CD81K04 complex where obtained at 20 °C in sitting drops by mixing 20 nl of protein complex solution with 20 nl of 20 % PEG 10k, 100 mM sodium acetate (pH 4.0) using acoustic liquid dispensing.Citation36 For crystals containing CD81K13 (scFv), 20% PEG3350, 200 mM sodium formate, 100 mM sodium citrate (pH 5.9) was used as precipitant solution. Crystals were prepared for flash cooling by adding glycerol to the crystallization drop solution to a final concentration of 20%.
Co-crystallization of Rb86 Fab fragment in complex with a pTau peptide (416-pS422–430)
Fab fragment at a concentration of 11.4 mg/ml in 10 mM Tris pH 7.4, 50 mM NaCl was incubated with pTau peptide (416-pS422–430) in a 5-fold molar excess for 3h at 21°C. Prior to crystallization experiments sodium acetate buffer at pH 4.5 was added to a final concentration of 0.2M followed by concentration of the protein to 20.7 mg/ml. Crystallization droplets were set up at 21 °C by mixing 0.1 μL of protein solution with 0.1 μL reservoir solution (Wizard1/2 Screen, Emerald) in vapor diffusion sitting drop experiments. Crystals were obtained out of 0.2 M lithium sulfate, 0.1 M sodium acetate and 30% PEG8000 as precipitant. Before data collection, crystals were transferred to crystallization buffer supplemented with 20% Glycerol and flash-frozen in liquid N2. Diffraction data were collected at a wavelength of 0.7000 Å using a PILATUS 6M detector at the beamline X10SA of the Swiss Light Source (Villigen, Switzerland).
Structure determination and refinement
The data were processed and scaled with XDS.Citation37 The structures were determined by molecular replacement with PHASER.Citation38 As search models, coordinates of in-house Fab and scFv structures were used. The coordinates were refined by rigid body and positional refinement with programs from the CCP4 suiteCitation39 and BUSTER.Citation40 Difference electron density was used to change amino acids according to the sequence differences, and to model the pTau peptide by real space refinement. Manual rebuilding of the proteins was done with COOT.Citation41 Data collection and refinement statistics can be found in Table S9 of the Supplemental Information.
Binding measurements
For the binding cell ELISA assay, HuH7-Rluc-H3, positive cell line expressing CD81, and HuH7-Rluc-L1, negative control cell line, were propagated in F-12 DMEM medium with 10% FCS at 37°C and 5% CO2. On day 1, the cells were trypsinized at approximately 90% confluence and resuspended at 4 × 105 cells/mL. Two x 104 cells/well HuH7-Rluc-H3 and HuH7-Rluc-L1 were plated in 50μL DMEM medium and allowed to adhere to the 96 well poly-D-Lysine plate for 24h at 37°C and 5% CO2. On day 2, the antibodies samples to be tested were prepared in a separate polypropylene round bottom plate with a twofold desired concentration with a final volume of 120 μl. All of the assay samples were diluted in cell culture medium. 50 μL of each antibody sample (duplicate wells) were added to cells to give final volume of 100 μL/well and incubated for 2 h at 4°C. Following the primary incubation the samples were removed by aspiration and the cells were fixed with 0.05% glutaraldehyde in PBS solution for 10 minutes at room temperature. After fixation, each well was washed 3 times with 200 μL PBS/0.05% Tween. The secondary incubation step for detection of bound anti-CD81 antibodies was performed for 2 h at room temperature on a reciprocal shaker. For the humanized CD81K04 and CD81K13 antibodies, detection was performed using peroxidase conjugate sheep anti-human-IgG-gamma chain specific antibody (The Binding Site Cat. # AP004) and a goat anti-mouse IgG, (H+L)-HRP conjugate BIORAD 170–6516 was used for the JS81 mouse positive control antibody (BD Biosciences 555675) both diluted 1:1000 in PBS 10% blocking buffer. Each well was washed 3 times with 200 μL PBS/0.05% Tween to remove unbound antibodies. The HRP activity was detected using 50 μL ready-to-use TMB solution and reaction was stopped after approximately 7–10 minutes with 50 μL per well 1M H2SO4. The absorbance was read using the ELISA Tecan reader at 450 nm with 620 nm reference wavelength.
For the rabbit antibody Rb86, we measured the supernatants at the first screening level for their ability to have association and dissociation parameters that do not deviate too much from the ones of the original antibody. The kinetic screening was performed on a BIAcore 4000 instrument, mounted with a BIAcore CM5 sensor.Citation42 A BIAcore CM5 series S chip was mounted into the instrument and was hydrodynamically addressed and preconditioned according to the manufacturer's instructions. The instrument buffer was HBS-EP buffer (10 mM HEPES (pH 7.4), 150 mM NaCl, 1 mM EDTA, 0.05 % (w/v) P20). An antibody capture system was prepared on the sensor surface. A polyclonal goat anti-human antibody with human IgG-Fc specificity (Jackson Lab.) was immobilized at 30 μg/ml in 10 mM sodium acetate buffer (pH 5) to spots 1, 2, 4 and 5 in the instrument's flow cells 1, 2, 3 and 4 at 10,000 RU using NHS/EDC chemistry. In each flow cell the antibodies were captured on spot 1 and spot 5. Spot 2 and spot 4 were used as reference spots. The sensor was deactivated with a 1 M ethanolamine solution. Humanized antibody derivatives were applied at concentrations between 44 nM and 70 nM in instrument buffer supplemented with 1mg/ml CMD (carboxymethyldextrane). The antibodies were injected at a flow rate of 30 μl/min for 2 min. The capture level (CL) of the surface-presented antibodies was measured in rel. response units (RU). The analytes in solution, phosphorylated human tau protein, non-phosphorylated human tau protein and the phosphorylated human tau mutant protein T422S, were injected at 300 nM for 3 min. at a flow rate of 30 μl/min. The dissociation was monitored for 5 min. The capture system was regenerated by a 1 min. injection of 10 mM glycine buffer pH 1.7 at 30 μL/min. over all flow cells. Two report points, the recorded signal shortly before the end of the analyte injection, denoted as binding late (BL) and the recorded signal shortly before the end of the dissociation time, stability late (SL), were used to characterize the kinetic screening performance. Furthermore, the dissociation rate constant kd (1/s) was calculated according to a Langmuir model and the antibody/antigen complex half-life was calculated in minutes according to the formula ln(2)/(60*kd). The binding late RU are compiled for each variant and for the reference rabbit antibody, as well as the dissociation rate constant kd [1/s]. For some variants that associated very poorly (RU in association phase close to zero or negative), it was not possible to determine a kd value. In these cases, the half-life value was set to zero.
Calculation of ABangle parameters and ABangle distances
The six ABangle orientation parameters (cp. ) of the crystal structures were calculated with the program code published by Dunbar et al.Citation21 available at http://www.stats.ox.ac.uk/∼dunbar/abangle. The program code was modified slightly so as to work with WolfGuy-numbered structures.
In order to compare similarity in ABangle space, we define a set of ABangle parameters as the tuple θ := (HL, HC1, LC1, HC2, LC2, dc) := (ϑ1, ϑ2, ϑ3, ϑ4, ϑ5, ϑ6).
The Euclidean distance between 2 sets of ABangle parameters is then As distABangle mingles angular (HL, HC1, LC1, HC2, LC2) with linear (dc) distance measures, it cannot be interpreted in terms of a factual unit of measure such as degrees.
ABangle predictor
The different ABangle parameters as described above were predicted by forest (multi-tree) recursive partitioning regression models implemented in Accelrys Pipeline Pilot 9.1.Citation43 The number of trees per forest was set to 200, and the maximum tree depth was set to 50.
The dataset for learning the predictor consisted of a redundant (with regard to Fv sequence) set of 1439 antibody Fv crystal structures that were crystallized as antibody-antigen complex at a resolution of a at least 3.0 Å. The data set of crystal structures was collected from the PDB (www.rcsb.org) in May 2015 and complemented by a small number of proprietary crystal structures owned by Roche. Fv structures crystallized in the absence of the antigen were not included as they were shown to have a higher perturbation in their VH-VL orientation parameters than Fv structures crystallized as a complex,Citation21 and thus do not contribute favorably to the predictor.Citation22
For each Fv structure involved, the ABangle parameters were measured and the orientation fingerprint consisting of 54 residues was generated.Citation44 To ensure to include the maximum diversity of different orientation fingerprints in the training set, we used CD-HITCitation45,46 to cluster the orientation fingerprints at 100% identity, and, for each cluster, added at least one representative to the training set, until ⅔ of the available structures had been assigned to the training set. The remaining ⅓ were used for testing. Due to the fact that the test set then consists of orientation fingerprints that are also included in the training set, the resulting Q2 values, ranging from 0.71 to 0.88 depending on the respective ABangle parameter, clearly overstate the actual capabilities of the predictor when confronted with an unknown orientation fingerprint. In that case, Q2 values were found to range 0.54 to 0.73, approximately.Citation22 To implement the leave-one-out predictions, the crystal structures of CD81K04, CD81K13 and Rb86, as well as any structures with the same orientation fingerprint (if any) were removed from the dataset for learning.
Statistical methods
Pearson correlation coefficient
The Pearson correlation coefficient measures the linear correlation between 2 variables X and Y. It is calculated as with cov(X,Y) being the covariance between X and Y and σ the standard deviation. We use the standard cor.test method in RCitation47 to calculate the correlation coefficient, and the p-value to evaluate if it differs significantly from zero.
RV coefficient
The RV coefficient was introduced by Escoufier to measure the similarity between square symmetric matrices.Citation32 The definition can be easily extended to rectangular matrices.Citation48 For 2 matrices X and Y, the RV coefficient can be calculated as with S = XXT and T = YYT. In order to calculate an associated p-value for the RV coefficient, we use the coeffRV-method from the FactoMineR packageCitation49 in R, which implements a permutation test as described in Josse et al.Citation50
Disclosure and potential conflicts of interest
AB, FL and GG are under paid employment by Roche Diagnostics GmbH. AK and JB are under paid employment by F. Hoffmann-La Roche AG. SH was under paid employment by Roche Palo Alto LLC when he generated the results presented in this article.
KMAB_A_1117720_supplementary_information.docx
Download MS Word (2.6 MB)Acknowledgments
We thank Johannes Auer, Steven Challand, and Michael Schräml for the cloning and expression of the humanized antibody variants and for the measurement of their binding properties. We are also grateful to Fiona Grüninger, Han Ma, Michael Brandt, Junjun Gao, and Ulrich Göpfert, who supported this work with their respective project. AB is funded by the Roche Postdoc Fellowship Program, which is hereby acknowledged.
References
- Brekke OH, Sandlie I. Therapeutic antibodies for human diseases at the dawn of the twenty-first century. Nat Rev Drug Discov 2003; 2:52-62; PMID:12509759; http://dx.doi.org/10.1038/nrd984
- Beck A, Wurch T, Bailly C, Corvaia N. Strategies and challenges for the next generation of therapeutic antibodies. Nat Rev Immunol 2010; 10:345-52; PMID:20414207; http://dx.doi.org/10.1038/nri2747
- Reichert JM. Marketed therapeutic antibodies compendium, mAbs 2012; 4:413-5; PMID:22531442; http://dx.doi.org/10.4161/mabs.19931
- Hansel TT, Kropshofer H, Singer T, Mitchell JA, George AJ. The safety and side effects of monoclonal antibodies. Nat Rev Drug Discov 2010; 9:325-38; PMID:20305665; http://dx.doi.org/10.1038/nrd3003
- Kabat EA, Te Wu T, Perry HM, Gottesman KS, Foeller C. Sequences of proteins of immunological interest. Darby PA: DIANE Publishing 1992
- Chothia C, Lesk AM. Canonical structures for the hypervariable regions of immunoglobulins. J Mol Biol 1987; 196:901-17; PMID:3681981; http://dx.doi.org/10.1016/0022-2836(87)90412-8
- Ahmadzadeh V, Farajnia S, Feizi MAH, Nejad RAK. Antibody humanization methods for development of therapeutic applications. Monoclon Antib Immunodiagn Immunother 2014; 33:67-73; PMID:24746146; http://dx.doi.org/10.1089/mab.2013.0080
- Safdari Y, Farajnia S, Asgharzadeh M, Khalili M. Antibody humanization methods -a review and update. Biotechnol Genet Eng Rev 2013; 29:175-86; PMID:24568279; http://dx.doi.org/10.1080/02648725.2013.801235
- Kuramochi T, Igawa T, Tsunoda H, Hattori K. Humanization and simultaneous optimization of monoclonal antibody. Methods Mol Biol 2014; 1060:123-37; PMID:24037839; http://dx.doi.org/10.1007/978-1-62703-586-6_7
- Almagro JC, Kodangattil S, Li J. Humanization of antibodies. In Making and Using Antibodies: A Practical Handbook, Second Edition. Editors Howard GC, Kaser MR 2013
- Gilliland GL, Luo J, Vafa O, Almagro JC. Leveraging SBDD in protein therapeutic development: antibody engineering. Methods Mol Biol 2012; 841:321-49; PMID:22222459; http://dx.doi.org/10.1007/978-1-61779-520-6_14
- Shirai H, Prades C, Vita R, Marcatili P, Popovic B, Xu J, Overington JP, Hirayama K, Soga S, Tsunoyama K, Clark D, Lefranc MP, Ikeda K. Antibody informatics for drug discovery. Biochim Biophys Acta 2014; 1844:2002-15; PMID:25110827; http://dx.doi.org/10.1016/j.bbapap.2014.07.006
- Kurella VB, Gali R. Structure guided homology model based design and engineering of mouse antibodies for humanization. Bioinformation. 2014; 10:180-6; PMID:24966517; http://dx.doi.org/10.6026/97320630010180
- Hanf KJ, Arndt JW, Chen LL, Jarpe M, Boriack-Sjodin PA, Li Y, van Vlijmen HW, Pepinsky RB, Simon KJ, Lugovskoy A. Antibody humanization by redesign of complementarity-determining region residues proximate to the acceptor framework. Methods 2014; 65:68-76; PMID:23816785; http://dx.doi.org/10.1016/j.ymeth.2013.06.024
- Kuroda D, Shirai H, Jacobson MP, Nakamura H. Computer-aided antibody design. Protein Eng Des Sel 2012; 25:507-21; PMID:22661385; http://dx.doi.org/10.1093/protein/gzs024
- Olimpieri PP, Marcatili P, Tramontano A. Tabhu: tools for antibody humanization. Bioinformatics 2015; 31:434-5; PMID:25304777; http://dx.doi.org/10.1093/bioinformatics/btu667
- Choi Y, Hua C, Sentman CL, Ackerman ME, Bailey-Kellogg C. Antibody humanization by structure-based computational protein design, mAbs 2015; 7(6):1045-57; PMID:26252731
- Marcatili P, Olimpieri PP, Chailyan A, Tramontano A. Antibody structural modeling with prediction of immunoglobulin structure (PIGS). Nat Protoc 2014; 9:2771-83; PMID:25375991; http://dx.doi.org/10.1038/nprot.2014.189
- Abhinandan KR, Martin AC. Analysis and prediction of VH/VL packing in antibodies. Protein Eng Des Sel 2010; 23:689-97; PMID:20591902; http://dx.doi.org/10.1093/protein/gzq043
- Chailyan A, Marcatili P, Tramontano A. The association of heavy and light chain variable domains in antibodies: implications for antigen specificity. FEBS J 2011; 278:2858-66; PMID:21651726; http://dx.doi.org/10.1111/j.1742-4658.2011.08207.x
- Dunbar J, Fuchs A, Shi J, Deane CM. ABangle: characterising the VH-VL orientation in antibodies. Protein Eng Des Sel 2013; 26:611-20; PMID:23708320; http://dx.doi.org/10.1093/protein/gzt020
- Bujotzek A, Dunbar J, Lipsmeier F, Schäfer W, Antes I, Deane CM, Georges G. Prediction of VH-VL domain orientation for antibody variable domain modeling. Proteins 2015; 83:681-95; PMID:25641019; http://dx.doi.org/10.1002/prot.24756
- Kaas Q, Ehrenmann F, Lefranc MP. IG, TR and IgSF, MHC and MhcSF: what do we learn from the IMGT Colliers de Perles? Brief Funct Genomics Proteom 2007; 6:253-64; http://dx.doi.org/10.1093/bfgp/elm032
- Niederfellner G, Lammens A, Mundigl O, Georges GJ, Schaefer W, Schwaiger M, Franke A, Wiechmann K, Jenewein S, Slootstra JW, et al. Epitope characterization and crystal structure of GA101 provide insights into the molecular basis for type I/II distinction of CD20 antibodies. Blood 2011; 118:358-67; PMID:21444918; http://dx.doi.org/10.1182/blood-2010-09-305847
- Vexler V, Yu L, Pamulapati C, Garrido R, Grimm HP, Sriraman P, Bohini S, Schraeml M, Singh U, Brandt M, et al. Target-mediated drug disposition and prolonged liver accumulation of a novel humanized anti-CD81 monoclonal antibody in cynomolgus monkeys, mAbs 2013; 5:776-86; PMID:23924796; http://dx.doi.org/10.4161/mabs.25642
- Ji C, Liu Y, Pamulapati C, Bohini S, Fertig G, Schraeml M, Rubas W, Brandt M, Ries S, Ma H, et al. Prevention of hepatitis C virus infection and spread in human liver chimeric mice by an anti-CD81 monoclonal antibody. Hepatology 2015; 61:1136-44; PMID:25417967; http://dx.doi.org/10.1002/hep.27603
- Collin L, Bohrmann B, Gopfert U, Oroszlan-Szovik K, Ozmen L, Gruninger F. Neuronal uptake of tau/pS422 antibody and reduced progression of tau pathology in a mouse model of Alzheimer disease. Brain 2014; 137:2834-46; PMID:25085375; http://dx.doi.org/10.1093/brain/awu213
- Kitadokoro K, Ponassi M, Galli G, Petracca R, Falugi F, Grandi G, Bolognesi M. Subunit association and conformational flexibility in the head subdomain of human CD81 large extracellular loop. Biol Chem 2002; 383:1447-52; PMID:12437138; http://dx.doi.org/10.1515/BC.2002.164
- Kitadokoro K, Bordo D, Galli G, Petracca R, Falugi F, Abrignani S, Grandi G, Bolognesi M. CD81 extracellular domain 3D structure: insight into the tetraspanin superfamily structural motifs. EMBO J 2001; 20:12-8; PMID:11226150; http://dx.doi.org/10.1093/emboj/20.1.12
- Krawczyk K, Baker T, Shi J, Deane CM. Antibody i-Patch prediction of the antibody binding site improves rigid local antibody-antigen docking. Protein Eng Des Sel 2013; 26:621-9; PMID:24006373; http://dx.doi.org/10.1093/protein/gzt043
- Mantel N. The detection of disease clustering and a generalized regression approach. Cancer Res 1967; 27:209-20; PMID:6018555
- Robert P, Escoufier Y. A unifying tool for linear multivariate statistical methods: the RV-coefficient. Appl Statist 1976:257-65; http://dx.doi.org/10.2307/2347233
- Jackson DA. PROTEST: a PROcrustean randomization TEST of community environment concordance. Ecoscience 1995:297-303
- Jayaram N, Bhowmick P, Martin AC. Germline VH/VL pairing in antibodies. Protein Eng Des Sel 2012; 25:523-9; PMID:22802295; http://dx.doi.org/10.1093/protein/gzs043
- Lloyd C, Lowe D, Edwards B, Welsh F, Dilks T, Hardman C, Vaughan T. Modelling the human immune response: performance of a 1011 human antibody repertoire against a broad panel of therapeutically relevant antigens. Protein Eng Des Sel 2009; 22:159-68; PMID:18974080; http://dx.doi.org/10.1093/protein/gzn058
- Villasenor AG, Wong A, Shao A, Garg A, Donohue TJ, Kuglstatter A, Harris SF. Nanolitre-scale crystallization using acoustic liquid-transfer technology. Acta Crystallogr D Biol Crystallogr 2012; 68:893-900; PMID:22868754; http://dx.doi.org/10.1107/S0907444912016617
- Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr 2010; 66:125-32; PMID:20124692; http://dx.doi.org/10.1107/S0907444909047337
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr 2007; 40:658-74; PMID:19461840; http://dx.doi.org/10.1107/S0021889807021206
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 2011; 67:235-42; PMID:21460441; http://dx.doi.org/10.1107/S0907444910045749
- Bricogne G, Blanc E, Brandl M, Flensburg C, Keller P, Paciorek W, Roversi P, Sharff A, Smart OS, Vonrhein C, et al. BUSTER version 2.11. Two. Cambridge, United Kingdom: Global Phasing Ltd 2011
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 2010; 66:486-501; PMID:20383002; http://dx.doi.org/10.1107/S0907444910007493
- Schräml M, Biehl M. Kinetic screening in the antibody development process. Methods Mol Biol 2012; 901:171-81; http://dx.doi.org/10.1007/978-1-61779-931-0_11
- Accelrys Software Inc. Pipeline Pilot, Release 9.1.0.13, San Diego: 2013
- Dengl S, Hoffmann E, Grote M, Wagner C, Mundigl O, Georges G, Thorey I, Stubenrauch KG, Bujotzek A, Josel HP, et al. Hapten-directed spontaneous disulfide shuffling: a universal technology for site-directed covalent coupling of payloads to antibodies. FASEB J 2015; 29:1763-79; PMID:25670234; http://dx.doi.org/10.1096/fj.14-263665
- Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006; 22:1658-9; PMID:16731699; http://dx.doi.org/10.1093/bioinformatics/btl158
- Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 2012; 28:3150-2; PMID:23060610; http://dx.doi.org/10.1093/bioinformatics/bts565
- Team RC R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, 2012, ISBN 3-900051-07-0 2012
- Abdi H. RV coefficient and congruence coefficient. Encyclopedia Measurement Statist 2007; 849-53
- Husson F, Josse J, Le S, Mazet J. FactoMineR: Factor Analysis and Data Mining with R. R package version 1.05. 2007; http://factominer.free.fr, http://www.agrocampus-rennes.fr/math
- Josse J, Pagès J, Husson F. Testing the significance of the RV coefficient. Comput Statist Data Anal 2008; 53:82-91; http://dx.doi.org/10.1016/j.csda.2008.06.012