
ABSTRACT
Tryptic peptide mapping analysis of a Chinese hamster ovary (CHO)-expressed, recombinant IgG1 monoclonal antibody revealed a previously unreported +16 Da modification. Through a combination of MSn experiments, and preparation and analysis of known synthetic peptides, the possibility of a sequence variant (Ala to Ser) was ruled out and the presence of hydroxylysine was confirmed. Post-translational hydroxylation of lysine was found in a consensus sequence (XKG) known to be the site of modification in other proteins such as collagen, and was therefore presumed to result from the activity of the CHO homolog of the lysyl hydroxylase complex. Although this consensus sequence was present in several locations in the antibody sequence, only a single site on the heavy-chain Fab was found to be modified.
Introduction
Recombinant monoclonal antibodies (mAbs) are a rapidly growing class of human therapeutics, and numerous products have been approved by health agencies or are currently under clinical investigation. A wide range of medical conditions have been successfully treated with therapeutic antibodies, including various forms of cancer,Citation1,2 a number of inflammatory disorders,Citation3,4 and other diseases.Citation5 In addition to conventional antibodies that contain a covalently linked tetramer of 2 full -length glycosylated heavy chains and 2 light chains, there are a number of other formats that have either been approved or are currently in clinical studies including glyco-engineered antibodies,Citation6 bispecific antibodies,Citation7 antibody-drug conjugates (ADCs),Citation8 and Fab fragments.Citation9 Regardless of the overall structure, manufacturing recombinant antibodies using bacterial, yeast or mammalian cell culture expression systems can lead to structural and chemical heterogeneity. DNA mutations leading to sequence variants of the recombinant product can be introduced during transfection or subsequent gene amplification.Citation10 The molecular machinery responsible for transcription and translation does not operate with 100% fidelity, and errors in these processes can result in various forms of microheterogeneity.Citation11 In addition, post-translational processing by host-cell enzymes can contribute additional complexity. Clone selection and cell culture conditions can be manipulated to influence levels of some post-translational modifications (PTMs), but detecting and controlling the levels of these modifications still remain major challenges for the biotechnology industry.Citation12,13
Peptide mapping by liquid chromatography-mass spectrometry (LC-MS) is an analytical tool commonly used to identify, characterize and quantify both expected and unexpected PTMs in therapeutic antibodies.Citation14 In a typical LC-MS peptide mapping experiment, an antibody is enzymatically digested with a highly specific enzyme such as trypsin to produce a set of peptides that can be studied further with MS. Since the sequence of a therapeutic antibody is known, as is the specificity of the enzyme used, the theoretical masses of the unmodified, and post-translationally modified peptides can be calculated with relative ease using bioinformatics tools.Citation15 A typical LC-MS experiment is designed to generate both intact peptide MS data, as well as MS/MS data, resulting from the isolation and subsequent fragmentation of precursor ions by collision-induced dissociation (CID). Thus, from a single experiment, it is possible to determine the occupancies of various modifications based on the extracted ion chromatogram (XIC) areas of both unmodified and post-translationally modified peptides, while also being able to confirm the site-specific identities of modifications or mutations based on the fragmentation data.
MS/MS data are typically interpreted with the assistance of bioinformatics tools by comparing observed fragment ion masses and intensities to those predicted by the software using the expected amino acid sequence of the protein.Citation15 It is also common for some degree of de novo sequencing to be necessary, especially when an unexpected post-translational modification is being considered, or when multiple interpretations are possible based on the observed fragment ions. Although complete peptide sequence coverage does not require a full ladder of b- and y-ions to be present in the MS/MS spectrum, confident localization of a modified residue can only be achieved if fragment ions adjacent to the modified residue are also observed. For example, a mass shift of ∼ +16Da can be due to oxidation of methionine (M) or tryptophan (W),Citation16 or due to a sequence variant (mutation) from alanine (A) to serine (S). If the initial MS/MS data are ambiguous, the observation of a neutral loss of 64Da (a unique fragment ion formed from oxidized M side chains) can help to distinguish oxidized M from oxidized W, and also from potential sequence variants.Citation17 Retention time differences can be another indicator of the identity of a modification, since oxidation alters the hydrophobicity of each residue to a different extent. In addition, orthogonal experiments can be performed to further elucidate the location of PTMs. Such an approach may utilize an enzyme with different cleavage specificity than the one used in the initial experiment, or one may design a different MS/MS fragmentation strategy targeting a potential modification. The fragmentation-based sequence coverage can be improved by strategically utilizing a targeted CID-based MSn design in an ion trap, by performing CID in a collision cell,Citation18 or by activating the precursor ions with another energy source (e.g., electron transfer dissociation, (ETD)Citation19,20) to provide different fragmentation selectivity.
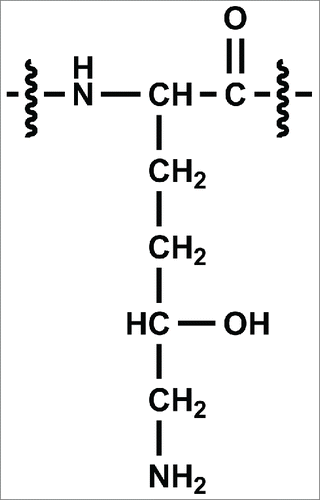
We report here the discovery of an unexpected post-translational modification, hydroxylation of lysine, in a Chinese hamster ovary (CHO)-expressed antibody. Lysine hydroxylation of collagen and proteins containing collagen-like domains occurs in animals and typically serves a functional/structural role as a precursor to crosslinking and O-glycosylation.Citation21,22 The hydroxylation of these lysines occurs via the lysyl hydroxylase enzyme, which recognizes the consensus amino acid sequence Xaa-Lys-Gly and converts lysine to 5-hydroxylysine (Hyl). The structure of Hyl is shown in . Although this modification is common in collagenous proteins, it has also been observed in some structurally unrelated proteins, such as the angler fish peptide hormone, somatostatin.Citation23 Furthermore, the presence of Hyl was previously reported in other biotherapeutic proteins derived from mammalian cells, including Activase® (r-tPA), a soluble form of CD4 receptor (rCD4), and a chimeric rCD4 variant (rCD4-IgG).Citation24 Each of these proteins was produced in cultured CHO cells, and the modification was found to only occur at lysine residues that were part of the Xaa-Lys-Gly consensus sequence. This specificity suggested that the proteins were modified by an endogenous lysyl hydroxylase enzyme. Although these proteins were unexpected substrates for lysyl hydroxylase, Hyl was found to have an occupancy ranging from 5 – 25% at certain consensus sequences. The work reported here suggests that a recombinant antibody may also be a substrate for the CHO homolog of this enzyme complex.
Figure 1. Chemical structure of 5-hydroxylysine (Hyl).
Results
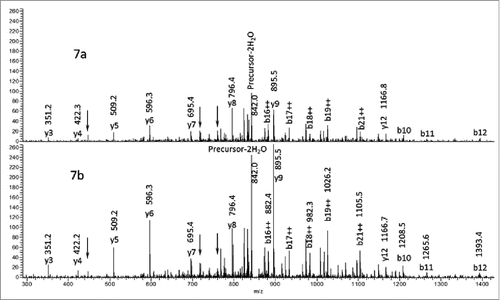
A CHO-expressed recombinant antibody, referred to here as mAb1, was characterized by tryptic peptide map analysis as described in the Materials and Methods section. An unknown peptide was observed with a mass corresponding to a +15.9948 Da (i.e., oxygen addition) modification on an expected tryptic peptide from the heavy-chain (HC101-HC124) of mAb1 with a sequence of XXXXXXXXXWGQGTLVTVSSASTK ([M+3H]3+= m/z 848.4116). shows the extracted ion chromatogram (XIC) of the modified (bottom panel) and unmodified (top panel) peptide forms. An extracted ion chromatogram is a signal trace where the intensity of ions from a defined m/z window is plotted versus retention time. The +15.9948 Da modified peptide (peak 3) of m/z 854.0789 [M+3H]3+ (2nd isotopic peak) was observed eluting slightly earlier than the unmodified version, indicating that the structural change decreased the hydrophobicity of the peptide. Two other minor peaks (1a and 1b) were also observed eluting before peak 3, which were determined to be isomers of the same peptide with the modification interpreted as an oxidation of the tryptophan residue. The amino acid position of the modification within the peptide for peak 3 was determined by analysis of the CID fragmentation data collected during a data-dependent acquisition MS/MS event for the modified peptide. This MS/MS event occurred slightly before the apex of the peak and is representative of the main species present at the selected precursor mass of the modified peptide. The MS/MS spectrum fragment ions labeled in bold, and with their m/z values, shown in narrowed down the location of the modification to the last 3 amino acids (STK), thus eliminating the possibility of oxidized tryptophan being the main component of the peak. The spectrum in suggests that the modification is localized to the 3 C-terminal residues (STK). The MS/MS spectrum of the unmodified peptide collected during the same LC-MS experiment is also displayed in , where the y3 ion is observed at 16 Da less than in the modified peptide. Based on this location of the +16 Da modification and the known literature, the highest probability considered was enzymatic hydroxylation of lysine. However, other possibilities were also evaluated.
Figure 2. XIC of modified and unmodified tryptic peptide XXXXXXXXXWGQGTLVTVSSASTK. (Note the ∼100-fold difference in intensity scale between upper and lower traces.) Peaks 1a and 1b correspond to tryptophan oxidation species that are isobaric with the modified peptide of peak 3.
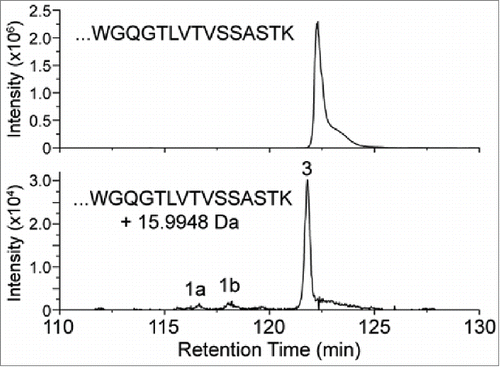
Figure 3. CID data from the modified (3a) m/z 854.1 [M+3H]3+ peptide and unmodified (3b) peptide m/z 848.8 [M+3H]3+. The oxygen addition is localized to one of the amino acids in bold.
![Figure 3. CID data from the modified (3a) m/z 854.1 [M+3H]3+ peptide and unmodified (3b) peptide m/z 848.8 [M+3H]3+. The oxygen addition is localized to one of the amino acids in bold.](/cms/asset/e99f58a7-d9cc-4053-98b2-a00db6718a0e/kmab_a_1122148_f0003_b.gif)
One modification that is isobaric to hydroxylation and was initially considered as a potential explanation for the +16 Da change is an alanine to serine (A121S) sequence variant. The synthetic peptide XXXXXXXXXWGQGTLVTVSSSSTK (S in bold and underlined indicates the position of the modification) representing the potential sequence variant was spiked into a sample of digested mAb1 that already contained a low level of the +16Da modification. shows the XIC of the sample monitored at m/z 854.0789 following addition of the synthetic peptide. The peak detected at 40.34 min (peak 2) was confirmed to be the synthetic peptide (MS/MS spectrum from peak 2 shown in Fig. S1 in Supporting Information) by MS/MS, and it was eluted as a new and distinct peak before the native +16 Da peak at 40.51 min (peak 3). Furthermore, peak 2 was only detected when this synthetic peptide was added. For example, it is not observed in the chromatogram shown in , where only the Hyl-containing synthetic peptide was spiked. The sequence of the A121S synthetic peptide was also confirmed by MS/MS during an LC-MS experiment in which the pure synthetic standard was analyzed without spiking into the mAb1 digest (see Supporting Information Fig. S2). The observation that the synthetic peptide containing the alanine to serine substitution elutes earlier than the modified peptide containing the oxygen addition suggests that the serine residue is not modified in mAb1, and provides further support for the hypothesis described above that the oxygen addition may be due to hydroxylation of the C-terminal lysine residue. As in , the peaks labeled 1a and 1b are W oxidation isomers of the native peptide. The peak with a retention time of 40.69 min (peak 4) is also identified to be W oxidation, but it is likely that this modification occurs post-column. The MS/MS spectra of peaks 1a, 1b, and 4 are shown in Supporting Information (Fig. S3). This interpretation is supported by the observation that the W oxidation species precisely co-elutes with the unmodified peptide. Furthermore, peaks 1a and 1b demonstrate that tryptophan oxidation results in earlier retention times compared to the other species, which indicates that the W oxidation identified in peak 4 was likely not present in the injected sample.
Figure 4. UPLC-MS extracted ion chromatogram of m/z 854.0789 [M+3H]3+ of tryptic digest from mAb1 spiked with a synthetic peptide containing alanine to serine sequence variant (identified as peak 2). The other identified peaks (1a , 1b and 4) correspond to peptides containing isomers of oxidized tryptophan at position 10 in the peptide, and the potential lysine to hydroxylysine modification at the C-terminus (peak 3).
![Figure 4. UPLC-MS extracted ion chromatogram of m/z 854.0789 [M+3H]3+ of tryptic digest from mAb1 spiked with a synthetic peptide containing alanine to serine sequence variant (identified as peak 2). The other identified peaks (1a , 1b and 4) correspond to peptides containing isomers of oxidized tryptophan at position 10 in the peptide, and the potential lysine to hydroxylysine modification at the C-terminus (peak 3).](/cms/asset/0cad147d-cfd5-4afe-b932-25dd36209431/kmab_a_1122148_f0004_b.gif)
The elution order of the potential PTMs leading to a net addition of one oxygen on the peptide XXXXXXXXXWGQGTLVTVSSASTK suggests that the +16 Da modification imparts a less significant change in hydrophobicity than the other modifications. In order of elution they are: (1a and 1b) Trp oxidation, (2) Ala to Ser and (3) native oxygen addition (i.e., potential Lys to Hyl). This observation suggests that the oxidation of a hydrophobic residue imparts a larger relative change to its hydrophobicity than oxidation of an already hydrophilic residue.Citation25
To definitively elucidate the location of the oxygen addition, an MSn experiment was conducted to provide additional fragmentation data from the STK residues at the C-terminus of the peptide. The CID MS/MS spectrum shown in was acquired via ion trap fragmentation, which is limited by a ∼1/3 low m/z cut off with the settings that were used.Citation26 The low m/z cutoff of this experiment means that all fragment ions formed with an m/z of ∼1/3 or less of that of the precursor ion will not be stable in the trap, and therefore not detected. The ∼1/3 cutoff of precursor ion m/z 854.1 [M+3H]3+precludes observation of b and y ions less than m/z 284.7, including the diagnostic y1 fragment ion at m/z 163.2. Due to this limitation, and the lack of detectable b-ions formed from fragmentation at the STK sequence, definitive confirmation of the modification site from the initial MS/MS experiment was not possible. To overcome this problem, MS/MS/MS spectra were collected in a subsequent experiment for both the unmodified and modified (+16 Da) forms of the tryptic peptides. In this experiment, the singly protonated y3 fragment ions at m/z 335.2 (unmodified) and m/z 351.2 (oxygen addition) were isolated from an MS/MS ion trap CID fragmentation event, and activated by CID again to acquire the MS/MS/MS spectra shown in . The y3 fragment ion was chosen as the precursor for the second stage of CID since it is composed of the 3 C-terminal residues (STK), where the oxygen addition was found in the MS/MS data. The b2 ion was observed in the spectra of modified and unmodified peptides, with m/z 189.1, corresponding to the m/z of a singly protonated b2 ion composed of unmodified Ser-Thr. This observation strongly suggests that neither Ser nor Thr residues contain the +16 Da modification. The m/z of the y1 ion observed from the unmodified peptide matches the predicted m/z of unmodified Lys, and is 16 Da less than the y1 ion observed for the modified peptide. Additionally, the MS/MS/MS spectrum of the modified peptide contains no y1 species corresponding to the m/z of unmodified Lys. Taken together, the MS/MS/MS data described above provides strong support that the +16 Da mass difference is located on the heavy chain lysine 124 residue.
Figure 5. MS3 of tryptic Hyl-containing (5a) and unmodified (5b) peptides. Precursor3+ (y3)1+ Fragments.
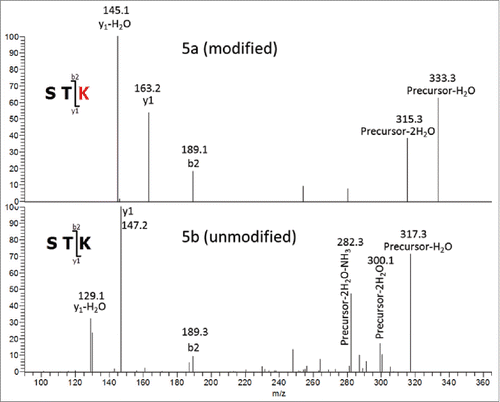
Figure 6. A portion of the extracted ion chromatograms (XICs) at m/z 854.0789 for the mAb1 tryptic digests both without (6a) and with (6b) addition of the synthetic Hyl-containing peptide. Peak 1c in each chromatogram corresponds to the retention times of both the native +16 Da peptide (6a) and the combined native and spiked Hyl-containing peptide (6b). Peaks 1a and 1b were identified as the oxidized tryptophan form of the peptide, which is isobaric with the Hyl-containing peptide.
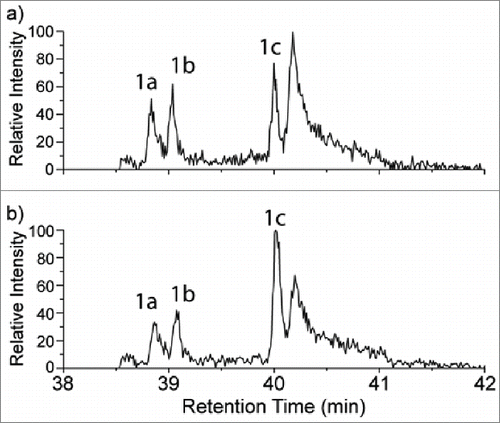
To verify the MS-derived assignment described above, the synthetic peptide containing 5-hydroxylysine at the proposed site was prepared by solid-phase synthesis and analyzed under the same LC-MS conditions as the mAb1 sample (see Supporting Information Fig. S2 for the MS/MS spectrum of the pure Hyl-containing synthetic peptide). The concentration of the endogenous +16 Da modified native peptide was calculated based on the tryptic digest concentration (500 µg/mL), mAb1 molecular weight (∼146 kDa) the site occupancy from %XIC (0.1%), and the molecular weight (2599.2Da) of the endogenous +16 Da modified native peptide (0.1%). Using these values, the absolute concentration of the +16 Da modified peptide was estimated to be 17.7 ng/mL in the digested sample. A 1-to-1 mixture of the synthetic and native +16 Da peptide was prepared by adding the synthetic peptide (1.77 µL @ 10µg/mL) into 1000 µL of the digest sample. This sample was analyzed by UPLC-MS so that both the LC retention times and the MS/MS fragmentation of the Hyl-containing synthetic and native +16 Da containing peptides could be compared in a single experiment. To improve the peak statistics of the low abundance peptides of interest, targeted analysis was performed for both samples with and without spiked addition of the synthetic peptide. A selected ion monitoring scan with an m/z range from 852.8 to 855.3 at high resolution (60,000 at 400 m/z) was followed by the targeted MS/MS full scan of m/z at 854.1 (2nd isotope) with an isolation width of 2.5 m/z. shows the XIC of the +16 Da-containing peptide in both the sample not spiked with the synthetic peptide (6a) and the spiked mAb1 digest sample (6b). The peak labeled 1c at a retention time of 40.0 min is present in both samples, and increases in intensity when the synthetic peptide is spiked. Furthermore, peak 1c remained as one peak with approximately the same peak width at the same retention time (0.02 min difference) following the spiked addition of the hydroxylysine-containing peptide. The co-elution of the synthetic, hydroxylysine-containing peptide and the endogenous +16 Da modified peptide derived from mAb1 provides further supporting evidence for the hydroxylysine assignment. The two peaks labeled as 1a and 1b are the W oxidation isomer peaks that were also described in .
shows stacked MS2 spectra of peak 1C in () and in () plotted on the same intensity scale to allow comparison. The spectra shown in this Figure correspond to the average of 6 scans for each sample. No new peaks were detected in the average MS2 spectrum in in comparison to the average MS2 spectrum in . In addition, there is a corresponding signal increase for all the labeled b and y ions in in comparison to . This result demonstrates the modified +16 Da peptide present in the digested mAb1 sample is identical to the synthetic Hyl-containing peptide standard. No intensity increase was observed for the 3 peaks labeled with black arrows, suggesting these peaks may be from a co-eluting interferent. The agreement between the 2 spectra provides further evidence supporting the interpretation that the mAb1 +16 Da modification is 5-hydroxylysine, consistent with the findings from the MSn experiments.
Figure 7. Comparison of the MS/MS spectra of peaks 1c in . Figure 7a corresponds to peak 1c in and Figure 7b corresponds to peak 1c in .
An interesting observation from the data reported here is that the modification of lysine to 5-hydroxylysine does not prevent tryptic cleavage. The tryptic peptide in which the modification was initially discovered contains the Hyl residue at its C-terminus, indicating that trypsin cleaved the peptide bond on the C-terminal side of this residue. Furthermore, the MS data were searched for the presence of the peptide containing the missed cleavage, in which Hyl was present as an internal residue, but it was not detected (data not shown). The uncleaved Hyl-containing peptide was synthesized to further study its susceptibility to tryptic cleavage. This peptide contained the Hyl residue internally, and the next Lys residue in the sequence at its C-terminus, with the rest of the sequence matching that of mAb1. This synthetic peptide described above was subjected to the same tryptic digestion procedure, and found to be completely cleaved at the Hyl residue (data not shown). These experiments are consistent with previous reports on tryptic selectivity for Hyl residues in collagenous proteins, and demonstrate the activity of trypsin at the corresponding position in mAb1.Citation27
Although the above data and literature demonstrate that trypsin cleaves on the C-terminal side of Hyl residues, it cannot be concluded from these data that trypsin necessarily cleaves Lys and Hyl with equal efficiency. An earlier paper reported up to a 7-fold reduction of susceptibility of Hyl to trypsin compared to unmodified Lys depending on the protein to enzyme ratio used during digest.Citation28 If trypsin cleaves Hyl less efficiently than Lys, this difference could affect the site occupancy determination by underestimating the Hyl levels (assuming the XICs of Hyl and Lys containing tryptic peptides are being compared). This possibility seems unlikely based on the data described above, in which no evidence of missed cleavage at the Hyl residue was found. Furthermore, the relative abundance of the unmodified miscleaved peptide compared to the fully cleaved peptide was found to be <1.0%. However, the levels of the Hyl-containing peptide are quite low (<1 %) with respect to the unmodified form, and it is possible that the missed cleavage form of the peptide is present but at levels too low to be detected. To help improve the confidence in the relative quantitation based on tryptic peptides, Asp-N and thermolysin digests were also performed. Neither of these enzymes depend on cleavage at the modified Lys residue. This selectivity allows for a relative quantitation experiment to be performed without potential bias based on differential cleavage rates at the modified and unmodified lysine. Each of these digests gave comparable levels of the 5-hydroxylysine containing peptide to those determined from trypsin (data not shown). The comparability of these peptide mapping data sets suggests that under the current digest conditions, trypsin completely cleaves both Hyl- and Lys-containing peptides.
Discussion
Hyl was found in a recombinant mAb, occurring at an XKG consensus sequence known to be recognized by lysyl hydroxylase. This result suggests that a mAb produced in CHO cells may be a substrate for the CHO homolog of lysyl hydroxylase. We have not yet observed the Hyl modification in mAbs produced in E. coli (unpublished data), where lysyl hydroxylase is absent. Interestingly, although mAb1 contains a total of 5 such consensus sequences in its heavy chain, hydroxylysine was found in only one of them by LC-MS analysis. Furthermore, no other lysine in mAb1 was found to contain the modification. One potential explanation that was considered was the possibility that the modified lysine residue was more solvent accessible than the other lysines. The solvent accessible surface areas (SASA) of several of the consensus sequence lysines were calculated using energy minimized structures by molecular dynamics with an AMBER force field.Citation29 While the SASA of the lysine side-chain at heavy-chain position 124 (i.e., the site where hydroxylation is observed) was found to be 92.6 Å2, the SASAs of 2 other positions, where no modification was evident by LC-MS, were actually found to be higher (122.1 and 130.7 Å2 at positions 43 and 65, respectively). Furthermore, the SASA of all lysine side-chains in mAb1 ranged from approximately 50 – 150 Å2, meaning that the modified residue is of ‘average’ solvent accessibility compared to other mAb1 lysines. Therefore, these data suggest that the presence of a solvent-accessible lysine, and a consensus sequence, are not the lone determinants for hydroxylation. One potential explanation for the lack of hydroxylysine at other consensus sequence positions is that the modification is not occurring via lysyl hydroxylase, and could be the result of another process such as chemical oxidation. This explanation, however, is highly unlikely. When mAb1 is subjected to a series of stress conditions, including strong visible light, AAPH (2,2′-azobis(2-amidinopropane) dihydrochloride), high temperature for extended times, low pH, and high pH, no change was observed to the levels of hydroxylysine at position 124, or the otherwise unmodified lysines (see Supporting Information Fig. S4). In addition, the hydroxylation of lysine due to chemical/environmental stress is unlikely given that the modification only occurs at one specific site, while other lysines are unmodified (even those that are more solvent accessible). A more plausible explanation for the specific modification at position 124 in the heavy chain is that the structure of mAb1 near this position may be a more favorable substrate for lysyl hydroxylase, whereas the local structures around other consensus sequence sites are not as readily bound by the enzyme. Targeted studies on the effect of residues nearby XKG consensus sequences in collagen have found strong correlations between the presence of specific amino acid residues and the likelihood of a site being a lysyl hydroxylase substrate.Citation30 In the case of the observed change to a mAb, it seems clear that solvent accessibility is not the only factor determining hydroxylation of XKG consensus sequence lysines.
In collagenous proteins it is common for Hyl to also be glycosylated to form an O-linked glycan (glucosyl-galactosylhydroxylysine).Citation31 These enzymatic modifications are catalyzed by a pair of enzymes, hydroxylysine galactosyltransferase and galactosylhydroxylysine glucosyltransferase, both of which are partners in the lysyl hydroxylase-3 complex that may also be responsible for the hydroxylation of lysine. Since these enzymes are known to exist together in a complex in mammalian cells, we considered the possibility that the glycosylated structure at heavy chain Lys124 could also exist. MS and MS/MS data collected during an LC-MS experiment on an Orbitrap Elite™ were searched for the presence of both the mono- and diglycoside structures. Since glycosylation is a more substantial modification than hydroxylation, we expected that trypsin would not likely cleave at the modified lysine (as it did for hydroxylysine, described above). Therefore, the MS and MS/MS data were searched for the modification by considering both the peptide including a missed tryptic cleavage at heavy-chain position 124, and by considering the peptide with cleavage at the modified residue. No definitive evidence was found for the presence of O-glycosylated hydroxylysine in mAb1.
In summary, an unexpected modification, 5-hydroxylysine, was found in a therapeutic mAb. This modification has been found to occur specifically on the heavy-chain at position 124, despite the presence of multiple lysyl hydroxylase consensus sequence sites. Several other CHO-expressed mAbs have been found to contain hydroxylysine at the same position (data not shown), implying that the modification may be fairly common in CHO-based antibodies. The modification was quantified and characterized by a variety of mass spectrometry-based methodologies. Although O-glycosylation at the Hyl residue was a possibility based on collagenous protein structures, no concrete evidence for this additional modification was found. Future research in collaboration with cell culture development will be needed to better understand the root cause of lysine hydroxylation in CHO cell expression of recombinant mAbs.
Materials and methods
The recombinant humanized IgG1 mAb was produced by transfected CHO cell lines and purified at Genentech (South San Francisco, CA). Trypsin was purchased from Roche (Indianapolis, IN). A BEH C18, 1.7µm 2.1 × 100 mm column was purchased from Waters Corporation (Milford, MA). A Jupiter C18 2.1 × 250 mm column was purchased from Phenomenex (Torrance, CA).
Peptide standards containing either a 5-hydroxylysine modification or an alanine to serine substitution (i.e., A121S) were synthesized internally using solid phase chemistry. Fmoc protected 5-hydroxylysine was purchased from PolyPeptide Labs (San Diego, CA). All other Fmoc-protected amino acids were purchased from Anaspec (Fremont, CA).
Peptide map procedure
Expressed antibodies were denatured and reduced with 6 M guanidine and 10 mM DTT at pH 8.1, 37° C for 1 hour. The denatured reduced protein was then buffer exchanged on a NAP-5 or PD-10 column (GE Healthcare Life Sciences, Pittsburgh, PA) into 25 mM TRIS, 1–2 mM CaCl2 at pH8.1 and digested separately by trypsin at 37° C for 4 hours or thermolysin at 25° C for 30 minutes. The digested samples were then analyzed by LC-MS/MS. Liquid chromatography was performed on a Waters Acquity H Class Bio UPLC (Waters Corporation, Milford, MA) or Agilent 1200 HPLC (Agilent Technologies, Santa Clara, CA) with a reversed-phase C18 column (Phenomenex, Torrance, CA or Waters, Milford, MA). The aqueous mobile phase (mobile phase A) contained 0.1% (v/v) trifluoroacetic acid (TFA) in HPLC grade water. The organic mobile phase (mobile phase B) contained 0.08% (v/v) TFA in acetonitrile. Two different gradients were utilized in this work: 1) UPLC experiments used a linear gradient of 7.5% to 42.5% mobile phase B over 52 min at 45°C; and 2) HPLC experiments used a column temperature of 55°C and a gradient of 0% to 35% mobile phase B over 140 min followed by an increase to 95% mobile phase B over 15 min followed by a re-equilibration step at 0% mobile phase B for 25 min. The HPLC/UPLC was coupled to an Orbitrap Elite™ mass spectrometer (Thermo Scientific, Waltham, MA). High resolution (60,000 at m/z 400) MS1 spectra were acquired with Orbitrap mass analyzers while low resolution MS/MS data were acquired in a Thermo Scientific LTQ™ ion trap mass spectrometer. The MS/MS event on the Orbitrap Elite™ was repeated for the top 5 precursor ions with dynamic exclusion enabled. The repeat count was set to 4 with a 30 second duration, and the exclusion duration for the precursor was set to 45 seconds. The resulting MS data were processed by Proteome Discoverer™ (Thermo Scientific), or Mascot Distiller (Matrix Science). Manual data interpretation was performed using Thermo Scientific's XCalibur™ software. GPMAW (Lighthouse data, Denmark) was used to calculate theoretical masses and the XIC was generated with the most intense isotope mass using a mass tolerance of + 10ppm.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KMAB_A_1122148_supplementary_material.docx
Download MS Word (282 KB)Acknowledgments
We thank Dr. Thomas Patapoff, Early Stage Pharmaceutical Development, Genentech Inc. for performing molecular dynamics calculations. We also thank Dr. Fred Jacobson, Protein Analytical Chemistry, Genentech Inc., for insightful discussions that contributed to this work.
Reference
- Scott AM, Wolchok, JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer 2012; 12:278-87; PMID:22437872; http://dx.doi.org/10.1038/nrc3236
- Beck A, Wurch T, Bailly C, Corvaia N. Strategies and challenges for the next generation of therapeutic antibodies. Nat Rev Immunol 2010; 10:345-52; PMID:20414207; http://dx.doi.org/10.1038/nri2747
- Andreakos E, Taylor PC, Feldmann M. Monoclonal antibodies in immune and inflammatory diseases. Curr Opin Biotechnol 2002; 13:615-20; PMID:12482523; http://dx.doi.org/10.1016/S0958-1669(02)00355-5
- Klotz L, Wiendl H. Monoclonal antibodies in neuroinflammatory diseases. Expert Opin Biol Ther 2013; 13:831-46; PMID:23521026; http://dx.doi.org/10.1517/14712598.2013.767329
- Desoubeaux G, Daguet A, Watier H. Therapeutic antibodies and infectious diseases, Tours, France, November 20-22, 2012. MAbs 2013; 5:626-32; PMID:23883703; http://dx.doi.org/10.4161/mabs.25300
- Goede V, Fischer K, Busch R, Engelke A, Eichhorst B, Wendtner CM, Chagorova T, de la Serna J, Dilhuydy MS, Illmer T, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med 2014; 370:1101-10; PMID:24401022; http://dx.doi.org/10.1056/NEJMoa1313984
- Bargou R, Leo E, Zugmaier G, Klinger M, Goebeler M, Knop S, Noppeney R, Viardot A, Hess G, Schuler M, et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 2008; 321:974-7; PMID:18703743; http://dx.doi.org/10.1126/science.1158545
- Chari RVJ, Miller ML, Widdison WC. Antibody-drug conjugates: an emerging concept in cancer therapy. Angew Chem Int Ed Engl 2014; 53:3796-827; PMID:24677743; http://dx.doi.org/10.1002/anie.201307628
- Rosenfeld PJ, Schwartz SD, Blumenkranz MS, Miller JW, Haller JA, Reimann JD, Greene WL, Shams N. Maximum tolerated dose of a humanized anti-vascular endothelial growth factor antibody fragment for treating neovascular age-related macular degeneration. Opthalmology 2005; 112:1048-53; http://dx.doi.org/10.1016/j.ophtha.2005.01.043
- Harris RJ, Murnane AA, Utter SL, Wagner KL, Cox ET, Polastri GD, Helder JC, Sliwkowski MB. Assessing genetic heterogeneity in production cell lines: Detection by peptide mapping of a low level Tyr to Gln sequence variant in a recombinant antibody. Nat Biotechnol 1993; 11:1293-7; http://dx.doi.org/10.1038/nbt1193-1293
- Liu H, Gaza-Bulsesco G, Faldu D, Chumsae C, Sun J. Heterogeneity of monoclonal antibodies. J Pharm Sci 2008; 97:2426-47; PMID:17828757; http://dx.doi.org/10.1002/jps.21180
- Walsh G, Jefferis R. Post-translational modifications in the context of therapeutic proteins. Nat Biotechnol 2006; 24:1241-52; PMID:17033665; http://dx.doi.org/10.1038/nbt1252
- Jeong KJ, Jang SH, Velmurugan N. Recombinant antibodies: engineering and production in yeast and bacterial hosts. Biotechnol J 2011; 6:16-27; PMID:21170983; http://dx.doi.org/10.1002/biot.201000381
- Houde D, Kauppinen P, Mhatre R, Lyubarskaya Y. Determination of protein oxidation by mass spectrometry and method transfer to quality control. J Chromatogr A 2006; 1123:189-98; PMID:16716331; http://dx.doi.org/10.1016/j.chroma.2006.04.046
- Zhang Z. Large-scale identification and quantification of covalent modifications in therapeutic proteins. Anal Chem 2009; 81:8354-64; PMID:19764700; http://dx.doi.org/10.1021/ac901193n
- Junyan JA, Zhang B, Cheng W, Yang YJ. Methionine, tryptophan, and histidine oxidation in a model protein, PTH: mechanisms and stabilization. J Pharm Sci 2009; 98:4485-500; PMID:19455640; http://dx.doi.org/10.1002/jps.21746
- Guan ZQ, Yates NA, Bakhtiar R. Detection and characterization of methionine oxidation in peptides by collision-induced dissociation and electron capture dissociation. J Am Soc Mass Spectrom 2003; 14:605-13; PMID:12781462; http://dx.doi.org/10.1016/S1044-0305(03)00201-0
- March RE. An introduction to quadrupole ion trap mass spectrometry. J Mass Spectrom 1997; 32:351-69; http://dx.doi.org/10.1002/(SICI)1096-9888(199704)32:4%3c351::AID-JMS512%3e3.0.CO;2-Y
- Wiesner J, Premsler T, Sickmann A. Application of electron transfer dissociation (ETD) for the analysis of posttranslational modifications. Proteomics 2008; 8:4466-83; PMID:18972526; http://dx.doi.org/10.1002/pmic.200800329
- Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci USA 2004; 101:9528-33; PMID:15210983; http://dx.doi.org/10.1073/pnas.0402700101
- Sricholpech M, Perdivara I, Yokoyama M, Nagaoka H, Terajima M, Tomer KB, Yanauchi M. Lysyl hydroxylase 3-mediated glucosylation in type I collagen. J Biol Chem 2012; 287:22998-3009; PMID:22573318; http://dx.doi.org/10.1074/jbc.M112.343954
- Siegal RC, Pinnell SR, Martin GR. Cross-linking of collagen and elastin. Properties of lysyl oxidase. Biochemistry 1970; 9:4486-92; PMID:5474144; http://dx.doi.org/10.1021/bi00825a004
- Andrews PC, Hawke D, Shively JE, Dixon JE. Anglerfish preprosomatostatin II is processed to somatostatin-28 and contains hydroxylysine at residue 23*. J Biol Chem 1984; 259:15021-4; PMID:6150931
- Molony MS, Wu S-L, Keyt LK, Harris RJ. The Unexpected Presence of Hydroxylysine in Non-Collagenous Proteins. Techn Protein Chem 1995; 6:91-8; http://dx.doi.org/10.1016/S1080-8914(06)80014-1
- Guo, DC, Mant CT, Taneja AK, Parker JMR, Hodges RSJ. Prediction of peptide retention times in reversed-phase high-performance liquid-chromatography I. Determination of retention coefficients of amino acid residues of model synthetic peptides. J Chromatogr A 1986; 359:499-518; http://dx.doi.org/10.1016/0021-9673(86)80102-9
- March RE, McMahon AW, Londry FA, Alfred RL, Todd JFJ, Vedel F. Resonance excitation of ions stored in a quadrupole ion trap. Part 1. A Simulation Study. Int J Mass Spectrom Ion Processes 1989; 95:119-56; http://dx.doi.org/10.1016/0168-1176(89)83037-X
- Wu GY, Pereyra B, Seifter S. Specificity of trypsin and carboxypeptidase-B for hydroxylysine residues in denatured collagens. Biochemistry 1981; 20:4321-4; PMID:7284327; http://dx.doi.org/10.1021/bi00518a013
- Molony MS, Quan C, Mulkerrin MG, Harris RJ. Hydroxylation of Lys residues reduces their susceptibility to digestion by trypsin and lysyl endopeptidase. Anal Biochem 1998; 258:136-7; PMID:9527859; http://dx.doi.org/10.1006/abio.1997.2513
- Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem 2004; 25:1157-74; PMID:15116359; http://dx.doi.org/10.1002/jcc.20035
- Risteli M, Niemitalo O, Lankinen H, Juffer AH, Myllyla R. Characterization of collagenous peptides bound to lysyl hydroxylase isoforms. J Biol Chem 2004; 279:37535-43; PMID:15208310; http://dx.doi.org/10.1074/jbc.M405638200
- Perdivara I, Perera L, Sricholpech M, Terajima M, Pleshko N, Yamauchi M, Tomer KB. Unusual fragmentation pathways in collagen glycopeptides. J Am Soc Mass Spectrom 2013; 24:1072-81; PMID:23633013; http://dx.doi.org/10.1007/s13361-013-0624-y