
ABSTRACT
Tumor necrosis factor (TNF)/TNF receptor (TNFR) superfamily members play essential roles in the development of the different phases of the immune response. Mouse LIGHT (TNFSF14) is a type II transmembrane protein with a C-terminus extracellular TNF homology domain (THD) that assembles in homotrimers and regulates the course of the immune responses by signaling through 2 receptors, the herpes virus entry mediator (HVEM, TNFSFR14) and the lymphotoxin β receptor (LTβR, TNFSFR3). LIGHT is a membrane-bound protein transiently expressed on activated T cells, natural killer (NK) cells and immature dendritic cells that can be proteolytically cleaved by a metalloprotease and released to the extracellular milieu. The immunotherapeutic potential of LIGHT blockade was evaluated in vivo. Administration of an antagonist of LIGHT interaction with its receptors attenuated the course of graft-versus-host reaction and recapitulated the reduced cytotoxic activity of LIGHT-deficient T cells adoptively transferred into non-irradiated semiallogeneic recipients. The lack of LIGHT expression on donor T cells or blockade of LIGHT interaction with its receptors slowed down the rate of T cell proliferation and decreased the frequency of precursor alloreactive T cells, retarding T cell differentiation toward effector T cells. The blockade of LIGHT/LTβR/HVEM pathway was associated with delayed downregulation of interleukin-7Rα and delayed upregulation of inducible costimulatory molecule expression on donor alloreactive CD8 T cells that are typical features of impaired T cell differentiation. These results expose the relevance of LIGHT/LTβR/HVEM interaction for the potential therapeutic control of the allogeneic immune responses mediated by alloreactive CD8 T cells that can contribute to prolong allograft survival.
Abbreviations
| LIGHT | = | homologous to lymphotoxin, exhibits inducible expression and competes with HSV glycoprotein D for binding to herpesvirus entry mediator, a receptor expressed on T lymphocytes |
| mAbM | = | monoclonal antibody |
| CD | = | Cluster of differentiation |
| HSV | = | Herpesvirus |
| TNF | = | Tumor necrosis factor |
| TNFR | = | Tumor necrosis factor receptor |
| HVEM | = | Herpesvirus entry mediator |
| LTβR | = | Lymphotoxin β receptor |
| CRD | = | Cysteine-rich domain |
| APC | = | Antigen-presenting cells |
| CTL | = | Cytotoxic T lymphocyte |
| NK | = | Natural killer |
| WT | = | Wild type |
| KO | = | Knock-out |
| Mgfp | = | monster green fluorescent protein |
| eGFP | = | enhanced green fluorescent protein |
| APC | = | Allophycocyanin |
| PE | = | Phycoerythrin |
| MHC | = | Major Histocompatibility Complex |
| PMA | = | Phorbol myristate acetate |
| CFSE | = | Carboxyfluorescein succinimidyl ester |
| FF-LIGHT | = | Flag-Foldon-tagged soluble mouse LIGHT |
| HVEM-Ig: HVEM.mIgG2a.Fc | = | Herpesvirus entry mediator bound to mouse IgG2a Fc fragment |
| LTβR-Ig: LTβR.huIgG1.Fc | = | Lymphotoxin beta receptor bound to human IgG1 Fc fragment |
Introduction
The tumor necrosis factor (TNF)/TNF receptor (TNFR) superfamily members play essential roles in diverse immunological processes such as T cell activation, costimulation, clonal expansion and T cell differentiation toward effector T cells.Citation1,2,3 The therapeutic intervention with biologics antagonizing the TNF/TNFR interactions can theoretically influence CD4 and CD8 T cell activation, clonal expansion, survival and particularly the process of differentiation toward effector T cells. This converts these interacting pathways as susceptible targets for the modulation of T cell-mediated immune responses.Citation4,5
Mouse LIGHT (TNFSF14) is a type II transmembrane protein of 239 amino acids with a C-terminal extracellular TNF homology domain (THD) that assembles as homotrimers capable to interact with HVEM Citation6 and LTβR.Citation7,8 There are 2 isoforms of mouse LIGHT produced by alternative splicing: an isoform without transmembrane domain that resides in the cytosol, and an isoform with a transmembrane domain that can be proteolytically processed at amino acid 84 to generate a soluble extracellular form of LIGHT.Citation6,9 In humans, there is an additional binding partner of LIGHT, a soluble protein named DcR3/TR6 protein (TNFRF6B) that lacks of transmembrane domain.Citation10,11
Mouse LIGHT displays a pattern of expression mainly restricted to activated T cells, NK cells and bone marrow immature dendritic cells.Citation6,12 Both LIGHT receptors, HVEM and LTβR, are expressed on haematopoietic and non-haematopoietic stromal cells, although the latter is not expressed on lymphoid cells.Citation13,14 Whereas LIGHT/HVEM main functional activity is a cosignaling pathway in T cells, LIGHT/LTβR interaction seems to be more relevant in regulating stromal/antigen-presenting cell (APC)/T cell cross-talk.Citation15
Preclinical studies in mouse models of disease are required to establish the proof of concept for the function of a target needed to start clinical studies in non-human primates and humans, which could then allow the use of new biologics for the treatment of immune-mediated diseases. However, high quality in vivo reagents to target the molecule of interest and its interactions are needed during the process. Our group has previously shown that antibody-mediated blockade of LIGHT/LTβR interaction with a partial antagonist antibody of LIGHT/LTβR interaction reduced short-term cytotoxic allogeneic responses, although it did not fully recapitulate the impaired cytotoxic response observed in LIGHT-deficient T cells.Citation12
Based on evidence gained in the field of transplantation using soluble LIGHT receptors as fusion proteins, such as HVEM.Ig and LTβR.Ig.Citation16,17,18 and from the phenotype of LIGHT-deficient mice,Citation19,12,17 we postulated that a complete blockade of LIGHT interaction with its receptors would contribute to achieve a more suitable pharmacological control of the allogeneic immune response. LIGHT blockade on T cells would impede its interaction with LTβR or HVEM on dendritic cells, and therefore hinder their maturation,Citation19,20,21,22 as well as prevent T/T cell collaboration through LIGHT/HVEM interactions that would contribute to maintenance of T cell survival during T cell expansion and differentiation.Citation23,24,25,26 To confirm this hypothesis, we characterized a set of anti-LIGHT antibodies raised in LIGHT-deficient mice and chose one that fully blocked the binding of soluble LTβR or HVEM to membrane LIGHT.
We demonstrated that efficient blockade of both HVEM/LIGHT and LTβR/LIGHT interactions attenuated the allogeneic immune response in a mouse model of graft-versus-host reaction and fully recapitulated the reduced cytotoxic phenotype of allogeneic LIGHT-deficient T cells. This study points to LIGHT as a suitable target for a better immunotherapeutic control of cytotoxic responses in transplantation.
Results
Clone 3D11 is a neutralizing anti-LIGHT antibody that efficiently blocks its receptor-binding site
To demonstrate the in vivo effects of LIGHT interaction with its receptors in the course of the allogeneic immune response, an antagonist mouse anti-mouse LIGHT monoclonal antibody (mAb; clone 3D11, mouse IgG2b, k) was raised in LIGHT-deficient mice immunized with LIGHT-transduced cells. The use of LIGHT-deficient mice as recipients for the immunization with LIGHT-transduced cells was critical for obtaining mouse anti-mouse LIGHT antibodies. LIGHT-deficient mice are not tolerant to LIGHT protein and they therefore can recognize LIGHT as a foreign antigen and mount a humoral immune response against multiple antigenic determinants of the protein.
The 3D11 antibody selectively bound NIH 3T3 cells expressing a green fluorescent protein (GFP): LIGHT fusion protein, in which GFP was fused to the intracellular N-terminus of mouse LIGHT, but did not recognize control NIH 3T3 cells expressing GFP only (, upper panel). The binding specificity was confirmed by pre-incubation of clone 3D11 with recombinant FF-LIGHT, which blocked the ability of 3D11 to bind LIGHT-transduced cells (, lower panel).
Figure 1. Effective blockade of LIGHT/LTβR and LIGHT/HVEM interactions using a mouse anti-mouse LIGHT monoclonal antibody. 2.5 × 105 NIH-3T3 cells transduced with GFP-tagged murine LIGHT (blue solid lines) or GFP-transduced NIH-3T3 cells (red solid lines) were incubated with a mouse anti-mouse LIGHT mAb 3D11. After an incubation step, antibody binding was revealed with biotinylated rat anti-mouse IgG2b followed by allophycocyanin-coupled streptavidin (A, upper panel). To further demonstrate the specificity of the anti-LIGHT mAb, 1 μg/well of Hilyte 647-labeled anti-LIGHT mAb alone (blue solid line) or 1 μg/well Hilyte 647-labeled anti-LIGHT mAb preincubated with 2 μg/well FF-LIGHT fusion protein (red solid line) were added to LIGHT-GFP transduced NIH 3T3 cells (A, lower panel). (B) 2.5 × 105 LIGHT-transduced NIH-3T3 cells were pre-incubated for 30 min at room temperature with anti-LIGHT mAb (clone 3D11, blue solid lines) or mouse IgG2b isotype control (red dotted lines). Then, cells were stained with LTβR-Ig (upper panel) or HVEM-Ig (lower panel). Binding of LTβR-Ig was revealed with biotinylated anti-hIgG and binding of HVEM-Ig was revealed with biotinylated anti-mouse IgG2a, both followed by allophycocyanin-coupled streptavidin. The baseline background staining is represented by hIgG1 Fc fragment (upper panel, black solid lines) to LIGHT-transduced NIH-3T3 cells or the binding of the Fc fragment mIgG2a (lower panel, black solid lines). The mean fluorescence intensity (MFI) is indicated in each plot. (C) Serial dilutions of anti-LIGHT mAb (clone 3D11) were preincubated with a fixed amount of 1 μg/well of soluble recombinant FF control (red solid line) or FF-LIGHT protein (black solid line). Then, 1 × 105 LIGHT-transduced NIH-3T3 were added to the reaction. After a washing step, a biotinylated rat anti-mouse IgG2b conjugate was further incubated and developed by SA-APC. The mean fluorescence intensity (MFI) binding of anti-LIGHT mAb to LIGHT transduced cells in the presence of FF control or FF-LIGHT is plotted. (D) The association and dissociation constant rates of 3D11 mAb binding to distinct immobilized mouse FcγR were calculated by surface plasmon resonance and from those values the equilibrium dissociation constant KD for each of them was determined.
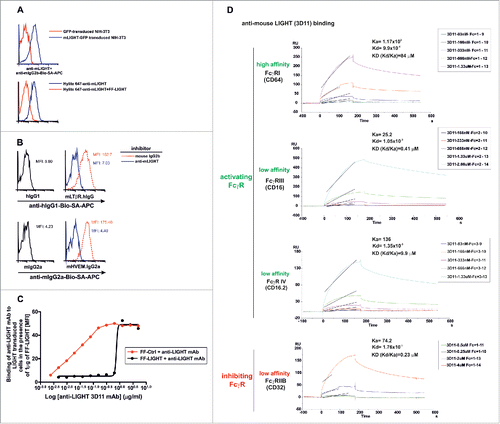
We next evaluated whether the anti-LIGHT mAb would prevent the interaction of mouse LIGHT expressed on cells with its receptors HVEM and LTβR. The 3D11 antibody antagonized the binding of sLTβR.Ig and sHVEM.Ig to mouse LIGHT-transduced cells down to background staining levels, similar to those obtained with isotype-matched control immunoglobulins (). The binding of anti-LIGHT mAb to LIGHT transduced cells was inhibited by FF-LIGHT at close to stoichiometry ratio assuming a monomeric antibody mass of 150 kDa and 2 recognition sites per antibody and 67.5 kDa for the FF-LIGHT trimer and 3 epitopes per molecule (at EC 50, 0.625 μg of antibody neutralized 1 μg FF-LIGHT) ().
We also assessed the binding avidity of the Fc fragment of anti-mouse LIGHT antibody to the activating and inhibiting mouse FcγR by surface plasmon resonance. The avidity constant (KD = Kd/Ka) for CD64 (FcγRI) and CD16.2 (FcγRIV) was 84 μM and 9.9 μM, whereas for CD16 (FcγRIII) and CD32 (FcγRIIB) was 0.41 μM and 0.23 μM, respectively (). This indicates that anti-LIGHT antibody (clone 3D11) binds preferentially to activating FcγRIII rather than FcγRIV or FcγRI.
We conclude from these results that 3D11 recognizes specifically mouse LIGHT but also fully inhibits the interaction of LIGHT with its receptors (HVEM and LTβR).
LIGHT protein expression is transiently detected upon polyclonal activation of T cells
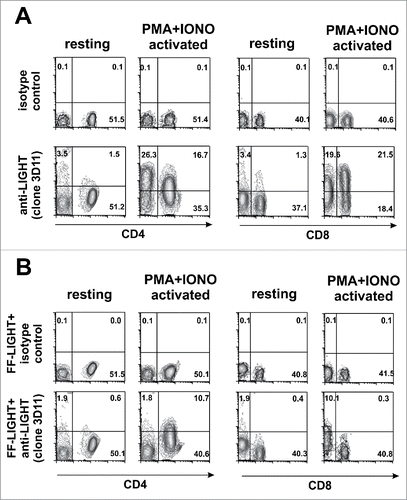
As it occurs for some other members of the TNF superfamily ligands such as CD40L or human LIGHT, its expression is only transient on activated T cells,Citation13,27 and visualization of this transient expression in vitro requires the presence of the fluorochrome-labeled antibody throughout the course of activation.Citation12 In C57BL/6 splenocytes stimulated for 5 h with PMA plus ionomycin, LIGHT expression was specifically detected with anti-LIGHT (3D11) antibody (), in line with results obtained previously with another anti-mouse LIGHT antibody.Citation12 Mouse LIGHT was only detected on activated CD4 and CD8 T cells, but not on resting T lymphocytes (). As expected for a specific staining, pre-incubation of the antibody with a molar excess of Flag-Foldon LIGHT abolished the staining of activated CD4+ and CD8+ T cells, respectively ().
Figure 2. Mouse LIGHT is rapidly upregulated on activated T cells after polyclonal stimulation. (A) Naïve C57BL/6 splenocytes (2 × 105) were left untreated or were stimulated with PMA (100 ng/ml) and ionomycin (500 ng/ml) for 5 h. 1 μg/well Hilyte-647-labeled anti mouse LIGHT (3D11) or Hylite-647-labeled isotype control mouse IgG2b were added to cultures during incubation in a final volume of 250 μl. A lineage cocktail to gate out CD19+ CD11c+ Ly6G+ cells was included in the staining. The expression of mouse LIGHT was then analyzed on live resting and activated CD4 and CD8 T cells. (B) The expression of mouse LIGHT on the surface of resting or PMA plus ionomycin activated CD4+ and CD8+ T cells was also determined with the anti-LIGHT antibody pre-incubated with Flag-Foldon LIGHT (FF-LIGHT) (2 μg FF-LIGHT per well). The percentage of each population is represented in each quadrant. PMA: Phorbol Myristate Acetate. Iono: Ionomycin.
Antibody mediated blockade of the LIGHT/LTβR/HVEM pathway recapitulates the attenuated cytotoxic allogeneic response of LIGHT-deficient T cells
LIGHT is required for lymph node hypertrophy in response to antigen immunization,Citation28 for T cell differentiation toward effector T cells in the course of an allogeneic immune responses Citation19,29 and for anti-tumor immunity.Citation18,30 Due to the lack of well-characterized anti-LIGHT antibodies, the therapeutic potential of modulating LIGHT has remained elusive because selective blockade of LIGHT cannot be properly achieved with soluble HVEM.Ig or LTβR.Ig fusion proteins that both bind several ligands.
With the novel anti-LIGHT antibody, we addressed in mice whether blockade of the LIGHT/LTβR/HVEM pathway could attenuate the course of graft-vs.-host reaction in a semiallogeneic adoptive transfer model of alloreactivity. To that aim, CB6F1 (F1) recipient mice were injected with either F1 splenocytes (control), or semiallogeneic B6 splenocytes (to induce graft-versus-host reaction), or LIGHT-deficient B6 splenocytes (to induce graft-vs.-host reaction in the absence of LIGHT on transferred cells). Mice adoptively transferred with semiallogeneic B6 splenocytes were treated with 3D11 (mouse anti-mouse LIGHT, isotype IgG2b) or with an isotype control mouse IgG2b. Nineteen days after the adoptive transfer, at the peak of the acute phase of the disease, the absolute number of host cells residing in the thymus, bone marrow and spleen was evaluated as a readout measurement of graft-versus-host reaction severity.Citation31,32 As expected, normal host cell numbers were found in F1 mice receiving F1 splenocytes, but almost all haematopoietic host cells were rejected 19 days after transfer of semiallogeneic B6 splenocytes. An equally efficient rejection of haematopoietic host cells was observed in the bone marrow, thymus (total and double-positive thymocytes) and spleen, i.e., in primary and secondary lymphoid tissues (). Transfer of LIGHT-deficient B6 splenocytes still lead to host cell rejection, but with reduced severity compared to wild type (p < 0.005), and the same was true when endogenous LIGHT was blocked with 3D11 upon transfer of wild type B6 splenocytes into F1 recipients (). No significant differences were found between the latter 2 conditions.
Figure 3. LIGHT blockade attenuated the course of graft-versus-host reaction in a mouse model of alloreactivity. Semiallogeneic splenocytes from C57BL/6 (70 × 106) were intravenously transferred into non irradiated CB6F1 recipients, which were treated with 1 mg of isotype mouse IgG2b control (red squares) or anti-LIGHT mAb (3D11, green triangles) at day 0. In a third experimental group, semiallogeneic splenocytes from LIGHT-deficient mice (70 × 106) were injected into CB6F1 mice (orange diamonds). The fourth group represents the syngeneic control group, in which 70 × 106 F1 splenocytes was injected into F1 recipients (blue circles). The absolute number of host bone marrow (A), thymocytes (B), double positive thymocytes (C) and splenocytes (D) was determined 19 days after the adoptive transfer. Data represent a pool of 3 independent experiments. Statistical significance is indicated as follows: *p < 0.05, **p < 0.005, ***p < 0.0005, and ns, non-significant.
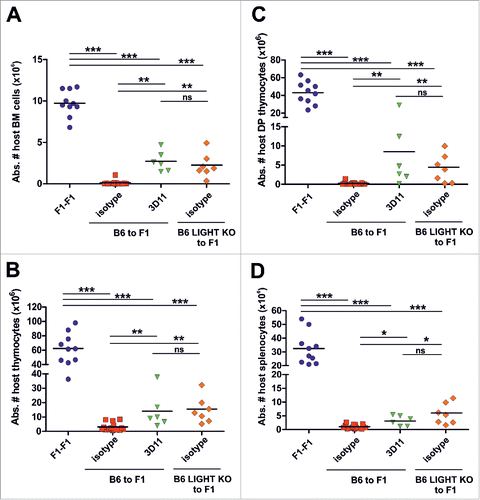
These results suggest that LIGHT blockade recapitulates the attenuated cytotoxic phenotype of LIGHT-deficient alloreactive T cells in F1 recipients, pointing to the relevance of LIGHT for the control of cytotoxic responses during the course of the allogeneic immune responses.
Blockade of LIGHT impairs allogeneic T cell proliferation almost as efficiently as its genetic ablation
To understand why LIGHT inhibition reduced the severity of the allogeneic cytotoxic response, we monitored the proliferative capacity of carboxyfluorescein succinimidyl ester (CFSE)-labeled donor alloreactive B6 splenocytes transferred into F1 recipient mice, in the presence or absence of genetic deficient LIGHT or immune therapeutic inhibition of LIGHT. Three days after the adoptive transfer of donor splenocytes, precursor frequencies (PF) and proliferation indexes (PI) were measured for CFSE-labeled donor CD4+ and CD8+ T cells (). The PF and PtdIns were significantly reduced in the presence of the anti-LIGHT antagonist antibody compared to treatment with the control antibody (). Moreover, the reduction achieved with the anti-LIGHT antibody was comparable to that obtained using LIGHT-deficient B6 donor cells ()
Figure 4. The proliferation index and frequency of donor CD4+ and CD8+ alloreactive T cells is altered after LIGHT blockade or in LIGHT-deficient T cells. (A-B) 70×106 of CFSE (carboxyfluorescein diacetate succinmidyl ester)-labeled B6 WT or CFSE-labeled B6 LIGHT-deficient splenocytes were adoptively transferred into non-irradiated F1 recipients and treated with 1 mg of isotype-control (mouse IgG2b) or mouse anti-mouse LIGHT (3D11) mAb at day 0. Three days later, the Proliferative Index (PI) and percentage of Precursor Frequency (PF) of donor alloreactive CD4+ and CD8+ T cells were determined using the ModFit LT software. Black line profile deconvoluted into cells that had divided once (green), twice (light violet), 3x (light blue), 4x (yellow), 5x (red), 6x (dark violet) and 7x or more (light green). X-axis represents CFSE fluorescence on a log scale and Y axis indicates cell counts (number of events). Data are representative of 2 independent experiments with 3 mice per group. Bars indicate mean ± SEM, and unpaired t test was used to compare differences between groups. Statistical significance was indicated as follows: *p < 0.05, **p < 0.005, ***p < 0.0005, and ns, non-significant.
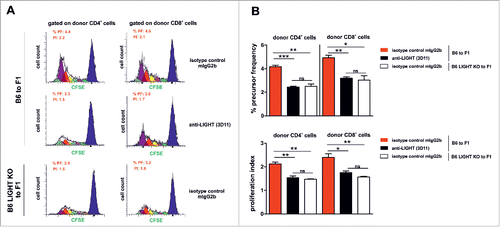
These results indicate that the functional blockade of LIGHT retards clonal expansion of alloreactive T cells.
LIGHT inhibition delays differentiation of alloreactive CD8+ T cells toward effector cells
Since LIGHT inhibition reduced proliferation indexes and precursor frequencies of donor alloreactive CD4+ and CD8+ T cells, we investigated whether T cell differentiation toward effector T cells had also been hampered. For this purpose, we monitored expression levels of costimulatory (BTLA, HVEM, inducible costimulatory molecule (ICOS)) and differentiation (interleukin (IL)-7Rα, KLRG-1) molecules on host and donor CD4+ and CD8+ T cells at 5 and 10 days after the semiallogeneic adoptive transfer of unfractionated parental B6 splenocytes into non-irradiated F1 recipient mice.
During a viral infection, CD8 T cell differentiation toward short-lived effector T cells and memory precursor cells can be tracked with IL-7Rα (CD127) and KLRG-1 surface markers.Citation33 IL-7Rα is the receptor for the homeostatic cytokine IL-7 whereas KLRG-1 is a membrane glycoprotein with a C-type lectin domain and one immunoreceptor tyrosine-based inhibitory motif (ITIM) expressed in NK subsets, effector and memory T cells.Citation33,34 KLRG-1 and CD127 (IL-7Rα) distinguish different stages of CD8 T cell differentiation at the peak of the response, just before the contraction phase, in which 95% of responding CD8 T cells are short-lived terminal effector cells (IL-7Rαlow KLRG-1hi).Citation33 KLRG-1 is, however, downregulated again during the contraction phase.Citation35 The expression of IL-7Rα was down-regulated on donor alloreactive CD4+ T cells in all experimental groups at day 5 after the adoptive transfer. This was also true for donor CD8+ T cells in the presence of LIGHT, but not for CD8+ T cells in conditions of genetic or pharmacologic impairment of LIGHT: in those cases, a fair percentage of IL-7Rα-positive CD8+ T cells was still observed at day 5, but these were gone at day 10 post transfer (, upper panel), suggesting that differentiation toward effector CD8 T cells was delayed in the absence of LIGHT.
Figure 5. Altered expression of differentiation (IL-7Rα and KLRG-1) and costimulatory (ICOS) surface markers is associated with delayed donor CD8 T cell differentiation in the absence of LIGHT. The pattern of expression of costimulatory and differentiation markers was assessed at days 5 and 10 after adoptive transfer of 70 × 106 of donor syngeneic or allogeneic B6 WT or B6 LIGHT-deficient splenocytes into F1 recipients. Recipients were treated on the day of the adoptive transfer with 1 mg of isotype control mouse IgG2b or anti-LIGHT mAb, clone 3D11. The expression of differentiation markers and costimulatory molecules was assessed by flow cytometry on host (blue lines) and donor (red solid lines) CD4+ and CD8+ T cells. The percentage of donor alloreactive CD8+ B6 WT or B6 LIGHT-deficient splenocytes that express IL7Rα or KLRG-1 (A-B) was evaluated 5 and 10 days after the adoptive transfer into F1 recipients. The delayed maturation of effector CD8+ T cells (ICOS negative) in the absence of LIGHT was also depicted (C-D). (E) The expression of the costimulatory markers BTLA and HVEM was also assessed at days 5 and 10. Bars indicate mean ± SEM. Statistical significance and p value was calculated using unpaired Student´s t test. The following criterion of significance was used: *p < 0.05, **p < 0.005, ***p < 0.0005, and ns, non-significant. Data are representative of 2 independent experiments with 3 mice per group.
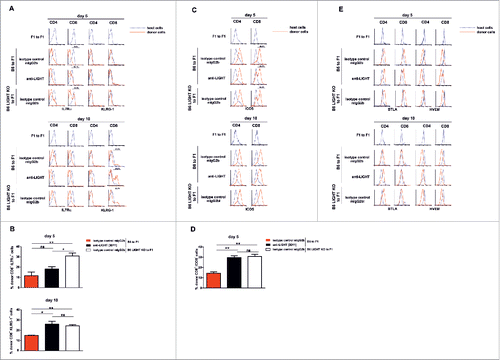
KLRG-1 expression was negative in all groups at day 5 post-adoptive transfer. At day 10, a KLRG-1-positive population of alloreactive CD8+ T cells emerged that was more prominent in experimental groups where LIGHT contribution was genetically or pharmacologically compromised (WT to F1, isotype control, 15.02% ± 0.16; WT to F1 anti-LIGHT 3D11; 25.96% ± 2.70; LIGHT knockout (KO) to F1, isotype control; 24.36% ± 0.96) (, lower panel). This could be interpreted as a delayed entry of CD8+ T cells in the contraction phase in the absence of LIGHT.
Unlike naïve T cells, which are ICOS negative, the expression of ICOS was upregulated at day 5 on most donor alloreactive CD8+ T cells in isotype-treated F1 mice. However, in the absence of LIGHT, there was still a fraction of ICOS− donor CD8 T cells at day 5 (14.48% ± 1.30 in isotype-treated controls vs. 29.63% ± 1.61 and 30.81% ± 2.12 in anti-LIGHT-treated and LIGHT-deficient donor cells, respectively), but not anymore at day 10 (), again pointing at delayed maturation of effector CD8+ T cells in the absence of LIGHT.
BTLA and HVEM expression were reciprocally regulated in donor CD4+ and CD8+ alloreactive T cells, after T cell activation and T cell expansion. Expression of BTLA is increased while that of HVEM was decreased. No significant changes in different surface markers were seen on donor alloreactive CD4+ or CD8+ T cells when experimental groups were compared to control ().
Overall, these results suggest that LIGHT is required on allogeneic CD8+ T cells to accelerate their differentiation.
Discussion
In the years to come, treatment of chronic and devastating diseases to which only palliative treatments are available will improve thanks to the substitution and complementation of chemical-based conventional therapy by biological-based approaches, including mAbs.Citation4,36,37 The understanding of how TNFSF and TNFRSF molecules contribute to the development of the immune response in different models of disease is essential for translation of the research findings into clinical practice. As members of the TNF/TNFRSF family control the absolute number of effector T cells and modulate the speed of the T cell differentiation process, they dictate the frequency of memory T cells that subsequently develop into long-lived memory T cells.Citation4,36
The process of T cell activation, costimulation, clonal expansion and differentiation toward effector T cells offers potential checkpoints for immune intervention. T cell activation with no or inefficient costimulation leads to functional inactivation, unresponsiveness or impaired T cell differentiation.Citation38,39,40 Therapeutics targeting the CD28/CTLA4/CD80/CD86 costimulatory pathway, which is dominated by members of the immunoglobulin superfamily, have had a major effect on the control of allogeneic responses. CTLA-4.Ig blockade of CD80 and CD86 interaction with CD28 and antibody-mediated blockade of CD40/CD40L are the 2 most successful therapeutic achievements for targeting costimulation in the field of preclinical transplantation.Citation41,42,43,44 CTLA4.Ig (belatacept) has already been introduced in the clinical setting as a maintenance treatment that efficiently prevents rejection while reducing the metabolic side effects of conventional drugs.Citation45 Treatment with CTLA4.Ig is particularly effective at low precursor frequency of alloreactive T cells that needs multiple rounds of division to reach a threshold effective to drive rejection.Citation46 However, CD8+ T cell-mediated rejection that is resistant to costimulation blockade remains an unbridgeable barrier, and thus alternative strategies aimed at targeting CD8 T cell differentiation that is independent of CD4 helper cells are required.Citation47,48
In a previous report with a partial antagonist of the LIGHT/LTβR interaction, short-term cytotoxic response was attenuated, although to a lesser extent than that seen in LIGHT-deficient mice.Citation12 Using a murine model of graft-versus-host reaction, in which host hematopoiesis is attacked by donor cytotoxic CD8 T cells, we now provide evidence that a fully antagonist antibody of LIGHT/HVEM and LIGHT/LTβR pathway, can down-modulate T cell responses to the same extent as that seen in LIGHT-deficient T cells. Since in vitro binding of LTβR to LIGHT competitively inhibits HVEM recognition of LIGHT when both receptors are expressed on the same cell, the blockade of LTβR/LIGHT by the anti-LIGHT antibody is likely to be more critical than blockade of HVEM/LIGHT. This accounts for the fact that this novel antibody (clone 3D11) fully recapitulates the phenotype of LIGHT-deficient T cells while a previously reported antibody (rat IgG2a anti-mouse LIGHT, clone 10F12) did not.Citation12
LIGHT has been proposed by several authors as a target for immunotherapy.Citation4,5,26,49,50,51 Nevertheless the development of a specific anti-LIGHT reagent has been complicated until recently due to the difficulty of generating active recombinant mouse LIGHT with productive binding affinity for its receptor and functional biological activity.Citation52 The description, functional evaluation and validation of this anti-LIGHT antibody opens up many possibilities to study the role of LIGHT and define its therapeutic potential in preclinical murine models of immune-related diseases (transplantation, tumor immunity and autoimmune diseases),Citation17,18,26,53,54,55 as well as in other pathologies in which LIGHT has been implicated, such as pulmonary fibrosis subsequent to chronic lung inflammation and idiopathic pulmonary fibrosis diseases,Citation56,57 skin fibrosis Citation58 and bone destruction through osteoclastogenesis.Citation59 This tool offers advantages over the classical approaches of using LTβR.Ig recombinant fusion protein because LTβR.Ig blocks LTαβ/LTβR in addition to LIGHT/LTβR/HVEM.Citation51 Administration of anti-LIGHT antibody in the above-mentioned disease models may help to clarify the contribution of LIGHT/LTβR/HVEM to the overall disease protection observed when using LTβR.Ig fusion protein. Conclusions drawn from results obtained with LTβR.Ig fusion proteins may need to be revisited because most studies utilized LTβR.Ig with a non-mutated Fc fragment of human IgG1 that binds with relatively high affinity to mouse FcγRIV, the main receptor implicated in antibody-dependent cell-mediated cytotoxicity (ADCC),Citation60,61 conferring this molecule with the potential to deplete cells expressing LTβR ligands.
In general, the mouse IgG2a isotype is far more efficient than either mouse IgG1 or IgG2b isotypes at mediating depletion through ADCC. The reason for this difference is because mouse IgG2a binds in vitro to all activating FcγRs (FcγRI>FcγRIV>FcγRIII), whereas mouse IgG2b binds mainly to FcγRIII and to a lesser extent to FcγRIV, but not to the high affinity activating receptor FcγRI. Moreover, the activating-to-inhibitory (A/I) ratio for mouse IgG2b (affinity of individual IgG subclasses for distinct activating FcγR and the inhibitory FcγRIIB) is higher than that of mouse IgG2a subclass, indicating that IgG2b binds with more avidity to inhibitory FcγRIIB than activating FcγR.Citation62 Monocytes express both FcγRI and FcγRIV whereas NK cells mainly express FcγRIII. Most in vivo ADCC depleting activity is mediated by monocytes (FcγRI and FcγRIV) rather than by NK cells (FcγRIII).Citation63 We could not demonstrate any in vitro ADCC or antibody dependent cell-mediated phagocytosis on opsonized LIGHT transduced target cells, and therefore it is very unlikely that the mouse IgG2b isotype of 3D11 hybridoma may mediate any in vivo ADCC-depleting activity. Besides, 3D11, an IgG2b subclass, exhibits a high binding avidity for the inhibitory FcγRIIB, and therefore a high A/I ratio. Co-ligation of IgG to inhibitory FcγRIIB receptor with high avidity reduces cellular responses mediated by activating receptors. The data suggest that 3D11 antibody may mediate in vivo effector function through neutralization of LIGHT interactions with HVEM and LTβR receptors rather than in vivo depletion through ADCC.
The experimental model used to dissect the role of LIGHT interaction with its receptors was the adoptive transfer of 70×106 of semiallogeneic B6 splenocytes into non-irradiated (recipient mice are not conditioned) F1 recipients (B6 × BALB/c). This large number of allogeneic splenocytes is required to overcome NK cell-mediated host resistance to engraftment of donor cells (hybrid resistance: resistance of host F1 to parental haematopoietic cell engraftment).Citation64 In this particular graft-vs.-host reaction model, the donor alloreactive lymphocytes eliminate host hematopoiesis starting at day 10, reaching the peak of rejection at around day 19. After this initial acute phase of rejection, recipient mice recover and parental engraftment starts to decline from week 3-4 post-adoptive transfer and disease very slowly progresses toward chronic inflammatory disease, reaching the peak at about 12 weeks after the semiallogeneic adoptive transfer. Although recipient hematopoiesis is practically abrogated at the acute phase of the disease (around day 19), no signs of gross pathology in the peripheral tissues are observed. Tissue infiltration requires the rupture of the epithelial barrier, and this does not occur in recipient F1 in which conditioning is not required for the implantation of donor T cells.Citation32
The adoptive transfer of parental B6 splenocytes into F1 recipients (BALB/c × B6) induces an allogeneic response mediated by donor CD8 cytotoxic cells that eliminate host hematopoiesis (graft-versus-host reaction). Donor CD4 T cells directly recognize host MHC class II alloantigens on host APC and provide cognate help to host B cells that experience transient expansion and autoantibody production. These activated B cells, as well as the host haematopoietic compartment, are soon attacked by activated B6 donor CD8 T cells that recognize host allogeneic MHC class I alloantigens and differentiate toward cytotoxic T cells with the help provided by donor B6 CD4 T cells.Citation32,65
In this mouse model, a reduced index of proliferation and precursor frequency of CD4 and CD8 T cells was observed after either pharmacological blockade or genetic ablation of LIGHT that correlated with impaired T cell expansion and differentiation toward effector T cells measured in an in vivo short-term assay of T cell proliferation tracked with CFSE. These observations were associated with a reduced cytotoxic activity against host F1 cells in a long-term in vivo assay evaluated at the peak phase of the acute graft-vs.-host reaction, 19 days after the adoptive transfer. This indicated that the cytotoxic activity of alloreactive T cells was reduced in the absence of LIGHT. These results are in line with the first phenotypic description of LIGHT KO T cells that denoted an alteration in allogeneic T cell proliferation in these mutant mice due to a defect on IL-2 secretion by CD4 T cells. This impaired production of IL-2 would perturb clonal expansion and the help provided to CD8 T cells to promote T cell division and differentiation toward effector T cells.Citation12,19
In the context of dendritic cell activation and maturation for proper costimulation, the majority of members of the TNFSF ligands (CD40L, LTαβ, LIGHT, TL1a, CD40L, OX-40L, and 4-1BBL), are molecules transiently expressed or highly upregulated upon T cell activation that would interact with the corresponding TNFR partner molecule on APC. These activated APCs would costimulate T cells, promote their division, survival and differentiation.Citation2 CD40L, along with LTαβ and LIGHT, are molecules transiently expressed in the early phase of T cell activation that differentially contribute to the licensing of dendritic cells through interaction with CD40 and LTβR respectively. LTαβ/LTβR and CD40/CD40L interactions appear to be the most relevant, while LIGHT provides a second layer of regulation.Citation20,21,22,66,67,68 Between LIGHT/LTβR and LIGHT/HVEM, the predominant interaction is the former, because LTβR outcompetes HVEM due its higher affinity for LIGHT.Citation12 Indirectly, LTβR signaling in stromal cells is also important for maintaining the integrity of lymphoid tissue, which is indispensable for the development of the immune response.Citation51,19,67 Indeed, LIGHT/LTβR interaction modulates lymph node hypertrophy by activating stromal cells expressing LTβR to release chemokines, upregulate adhesion molecules and thus attract tissue-derived dendritic cells to the draining lymph nodes.Citation28
IL-7Rα and KLRG-1 are reciprocally modulated on CD8 T cells as they differentiate toward effector CD8 T cells in viral infection models. Combination of these 2 markers permits the process of T cell differentiation to be followed.Citation33,69 In our hands, the use of bright fluorochromes attached to anti-mouse CCR7 antibody (clone 4B12) in combination with CD62L gave a weak staining that could not clearly discriminate the different stages of T-cell differentiation. The absence of LIGHT (LIGHT-deficient T cells or antibody blockade of LIGHT) leads to delayed downregulation of IL-7Rα and delayed upregulation of ICOS cells at day 5 after the adoptive transfer, suggesting that T cell differentiation was hampered in donor alloreactive CD8 T cells in the late phase of T cell activation and clonal expansion. Moreover, T-cell differentiation toward effector cells is associated with an increased percentage of KLRG-1high short-lived effector cells (SLECs) in the allogeneic adoptively transferred CD8 T cells, whereas the initiation (memory precursor cells) and the contraction phase (long-lived memory cells) is characterized by downregulation of KLRG-1.Citation35 The augmented percentage of KLRG-1high SLECs in the absence of LIGHT suggests a defect in T cell differentiation at day 10, which is linked to a less effective clonal expansion at day 3 after the adoptive transfer.
In summary, the data presented here provide evidence for the therapeutic use of LIGHT inhibitors to dampen immune responses via attenuation of donor allogeneic CD8 T cell responses.
Material and methods
Animals
Eight to 12 weeks-old female C57BL/6 (H-2b, B6, CD45.2), BALB/c (H-2d), CB6F1 (BALB/c × C57BL/6 F1) mice (H-2d/b) and LIGHT-deficient mice (LIGHT−/−) backcrossed more than 15 generations onto C57BL/6 background were bred at the animal facility of the University of Leon (Spain).Citation19 All experiments with rodents were performed in accordance and following animal protocols specifically approved for the Ethical Committee for Animal Research of the School of Veterinary Medicine (University of Leon), the Animal Welfare Committee of University of Alcala de Henares (Madrid) and followed the European Guidelines for Animal Care and Use of Laboratory Animals.
Monoclonal antibodies and surface plasmon resonance
LIGHT-deficient mice were immunized intraperitoneally with 0.25 ml of a 1:1.2 mixture of 5-10 × 106 of GFP-tagged mouse LIGHT transduced into NIH-3T3 cells in Freund's Incomplete Adjuvant (Sigma).Citation12 Six weeks after the priming immunization, mice received an intravenous booster injection of LIGHT-transduced cells in saline. The immortalization of mouse B cells with myeloma cell line X63 Ag8.653 was previously described Citation70 and supernatants of the heterohybridomas secreting rat mAbs were tested 10 days after for their specificity against LIGHT transduced cells by flow cytometry.
Flag-Foldon-tagged soluble mouse LIGHT (FF-LIGHT),Citation71 soluble FF-control protein (FF-Ctrl) and LTβR.human IgG1.Fc (LTβR.Ig) were produced and purified as previously reported.Citation72 Recombinant mouse HVEM.mouse IgG2a.Fc (HVEM.Ig) produced in insect cells was a gift from Genentech. Commercially available mouse IgG2a and human IgG1 were used as matched isotype control for the recombinant proteins.
For the screening of anti-LIGHT antibodies displaying antagonist activity and therefore capable to prevent LIGHT interaction with its receptors, 2.5 × 105 LIGHT-transduced NIH-3T3 cells were incubated with a saturating amount of isotype matched control or anti-mouse LIGHT mAbs for 30 min at room temperature (2 μg/ml). In the presence of competitor antibody, cells were then further incubated for 2 h at 37°C with an optimal dilution of either HVEM-Ig (5 μg/well) or LTβR-Ig (1 μg/well) in a final volume of 100 μl. After a washing step, the reaction was developed with the appropriate biotinylated conjugates (biotinylated rat anti-mouse IgG2a isotype specific mAb, clone R19-15, BD Biosciences or mouse anti-human IgG Fc fragment, Jackson ImmunoResearch), followed by allophycocyanin-coupled to streptavidin.
The BIACORE 3000 system, sensor chip CM5, surfactant P20, amine coupling kit containing N-hydroxysuccinimide (NHS) and N-Ethyl-N'-dimethylaminopropyl carbodiimide (EDC), were from BIACORE (Upsala, Sweden). All biosensor assays were performed with HEPES-buffered saline (HBS-EP) as running buffer (10 mM HEPES, 150 mM sodium acetate, 3 mM magnesium acetate, 3.4 mM EDTA and 0.005% surfactant P20, pH 7.4). The different compounds were dissolved into running buffer.Citation73
FcγRI (CD64), FcγRIIB (CD32), FcγRIIIA (CD16) and FcγRIV (CD16.2) were immobilized at 50 μg/ml in formate buffer, pH 4.3 by injection onto the EDC/NHS-activated surface of a CM5-type sensor chip until a signal of approximately 6000 RU was obtained. Free activated sites of the matrix were saturated by injection of 20 μL ethanolamine hydrochloride, pH 8.5. All the binding experiments were carried out at 25°C with a constant flow rate of 30 μl/min. Different concentrations of mouse anti-LIGHT (clone 3D11) were injected for 3 min followed by a dissociation phase of 3 min. The sensor chip surface was regenerated after each experiment by injection of 20 μl of 10 mM NaOH.
The kinetic parameters were calculated using the BIAeval 4.1 software on a personal computer. Global analysis was performed using the simple Langmuir binding model. The specific binding profiles were obtained after subtracting the response signal from the channel control (ethanolamine) and from blank buffer injection. The fitting to each model was judged by the reduced chi square and randomness of residue distribution.
Flow cytometry
Mouse anti-mouse LIGHT (3D11, mouse IgG2b) and mouse IgG2b isotype control (MPC-11, Biolegend) mAbs were labeled with Hilyte-Fluor 647 according to the manufacturer's protocol (Anaspec Inc.). CD4 (GK1.5), CD8α (53-6.7), Ly6G (1A8), CD19 (6D5), ICOS (7E.17G9), CD127 (IL-7Rα, A7R34), and killer cell lectin-like receptor subfamily G, member 1 (KLRG-1, MAFA) mAbs were all purchased from Biolegend (USA). To distinguish haematopoietic cells of donor parental B6 and host F1, cells from distinct haematopoietic compartments were stained with FITC-conjugated anti–H-2d (SF1-1.1) and Alexa 647-conjugated anti-H-2b (AF6-88.5). Rat mAb anti-mouse BTLA (CD272, clone 4G12b) Citation74 and anti-mouse HVEM (CD250, clone 10F3) Citation29 were produced and labeled in-house and used in this study.
Fc gamma receptors were blocked by incubating cell suspensions with 2 μg/ml of blocking anti-FcγR (rat IgG2b anti-FcγRII/III mAb, clone 2.4G2) before staining with fluorochrome-labeled antibodies.Citation75 Dead cells were excluded from the acquisition gate by staining with propidium iodide. Samples were acquired on a Cyan 9 cytometer (Beckman Coulter, Miami, FL, USA) and data analysis was performed using WinList version 7.0 (Verity Software House, Topsham, ME, USA).
In vitro polyclonal T cell activation to induce LIGHT expression
Naïve C57BL/6 splenocytes were polyclonally activated in vitro with Phorbol Myristate Acetate (PMA, 100 ng/ml) plus ionomycin (500 ng/ml) or were left untreated for 5 h at 37°C. The transient expression of LIGHT was analyzed on resting and polyclonally activated T cells (2 × 105 cells / well) stained with 1 μg/well of Hylite 647-labeled anti-LIGHT mAb (clone 3D11) or Hylite 647-labeled isotype-matched mouse IgG2b (MPC-11) during the time of incubation, using a lineage cocktail to gate out all cells with phagocytic phenotype (CD19+ Ly6G+ CD11c+) that take up the antibody unspecifically.
In vivo proliferative assay of CFSE-labeled donor alloreactive T cells
70 × 106 of B6 WT or LIGHT-deficient unfractionated splenocytes were labeled with 5 μM carboxyfluorescein diacetate succinmidyl ester (CFSE) and adoptively transferred into non-irradiated F1 recipients, according to Lyons et al.Citation76 The day of the adoptive transfer, recipient mice were treated with 1 mg (˜40 mg/kg) of isotype-control (mouse IgG2b) or mouse anti-mouse LIGHT (3D11) mAb. Three days later, the PI and PF of alloreactive CD4+ and CD8+ T cells were determined by deconvoluting and analyzing the reduced CFSE fluorescence using the ModFit LT 4.1 version (Verity software, ME). PtdIns, is a measure of the increase in cell number over the course of the assay. PF returns the fraction of cells in the initial population that responded to the stimulus by proliferating. The location of each generation of cells is represented by a unique peak color.
In vivo murine model of Graft-versus-Host Reaction
In a third experimental group, F1 recipient mice were inoculated with 70 × 106 splenocytes from LIGHT-deficient mice in B6 background and treated with 1 mg isotype control. Finally, a syngeneic control group was included in which 70 × 106 syngeneic F1 splenocytes were injected into non-irradiated F1 recipients. The absolute number of haematopoietic cells in primary and secondary lymphoid organs was recorded 19 days after the adoptive transfer.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Statistical analysis
Collected data from experimental and control groups were analyzed using Graph Pad prism Version 5. Statistical significance was assessed using the non-parametric Mann-Whitney t test or Student´s t test with Welch´s correction to compare the means among groups. A p value less than 0.05 was considered statistically significant.
Funding
This work has been supported by grants of the Spanish Ministry of Health (Fondo de Investigaciones Sanitarias, PI13/00029), Department of Education of Castilla and Leon Regional Government (Grant# LE093U13) and Mutua Madrileña Foundation (Basic research grants 2012) to J.I.R.B; by Miguel Servet National Grant (Health National Organization Research Program) CP12/03063 and by Gerencia Regional de Salud GRS963/A/2014 and GRS1142/A/2015 to M.L.R.G; and by the Swiss National Science Foundation to PS.
References
- Croft M. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol 2009; 9:271-85; PMID:19319144; http://dx.doi.org/10.1038/nri2526
- Croft M. The TNF family in T cell differentiation and function–unanswered questions and future directions. Semin Immunol 2014; 26:183-90; PMID:24613728; http://dx.doi.org/10.1016/j.smim.2014.02.005
- Sedy J, Bekiaris V, Ware CF. Tumor Necrosis Factor Superfamily in Innate Immunity and Inflammation. Cold Spring Harb Perspect Biol 2014; 7:a016279; PMID:25524549
- Croft M, Benedict CA, Ware CF. Clinical targeting of the TNF and TNFR superfamilies. Nat Rev Drug Discov 2013; 12:147-68; PMID:23334208; http://dx.doi.org/10.1038/nrd3930
- Ware CF. Targeting lymphocyte activation through the lymphotoxin and LIGHT pathways. Immunol Rev 2008; 223:186-201; PMID:18613837; http://dx.doi.org/10.1111/j.1600-065X.2008.00629.x
- Mauri DN, Ebner R, Montgomery RI, Kochel KD, Cheung TC, Yu GL, Ruben S, Murphy M, Eisenberg RJ, Cohen GH, et al. LIGHT, a new member of the TNF superfamily, and lymphotoxin α are ligands for herpesvirus entry mediator. Immunity 1998; 8:21-30; PMID:9462508; http://dx.doi.org/10.1016/S1074-7613(00)80455-0
- Rooney IA, Butrovich KD, Glass AA, Borboroglu S, Benedict CA, Whitbeck JC, Cohen GH, Eisenberg RJ, Ware CF. The lymphotoxin-β receptor is necessary and sufficient for LIGHT-mediated apoptosis of tumor cells. J Biol Chem 2000; 275:14307-15; PMID:10799510; http://dx.doi.org/10.1074/jbc.275.19.14307
- Zhai Y, Guo R, Hsu TL, Yu GL, Ni J, Kwon BS, Jiang GW, Lu J, Tan J, Ugustus M, et al. LIGHT, a novel ligand for lymphotoxin β receptor and TR2/HVEM induces apoptosis and suppresses in vivo tumor formation via gene transfer. J Clin Invest 1998; 102:1142-51; PMID:9739048; http://dx.doi.org/10.1172/JCI3492
- Granger SW, Butrovich KD, Houshmand P, Edwards WR, Ware CF. Genomic characterization of LIGHT reveals linkage to an immune response locus on chromosome 19p13.3 and distinct isoforms generated by alternate splicing or proteolysis. J Immunol 2001; 167:5122-8; PMID:11673523; http://dx.doi.org/10.4049/jimmunol.167.9.5122
- Zhang J, Salcedo TW, Wan X, Ullrich S, Hu B, Gregorio T, Feng P, Qi S, Chen H, Cho YH, et al. Modulation of T-cell responses to alloantigens by TR6/DcR3. J Clin Invest 2001; 107:1459-68; PMID:11390428; http://dx.doi.org/10.1172/JCI12159
- Yu KY, Kwon B, Ni J, Zhai Y, Ebner R, Kwon BS. A newly identified member of tumor necrosis factor receptor superfamily (TR6) suppresses LIGHT-mediated apoptosis. J Biol Chem 1999; 274:13733-6; PMID:10318773; http://dx.doi.org/10.1074/jbc.274.20.13733
- del Rio ML, Fernandez-Renedo C, Scheu S, Pfeffer K, Shintani Y, Kronenberg M, Chaloin O, Schneider P, Rodriguez-Barbosa JI. Therapeutic blockade of LIGHT interaction with herpesvirus entry mediator and lymphotoxin β receptor attenuates in vivo cytotoxic allogeneic responses. Transplantation 2014; 98:1165-74; PMID:25226173; http://dx.doi.org/10.1097/TP.0000000000000417
- Morel Y, Schiano de Colella JM, Harrop J, Deen KC, Holmes SD, Wattam TA, Khandekar SS, Truneh A, Sweet RW, Gastaut JA, et al. Reciprocal expression of the TNF family receptor herpes virus entry mediator and its ligand LIGHT on activated T cells: LIGHT down-regulates its own receptor. J Immunol 2000; 165:4397-404; PMID:11035077; http://dx.doi.org/10.4049/jimmunol.165.8.4397
- Force WR, Walter BN, Hession C, Tizard R, Kozak CA, Browning JL, Ware CF. Mouse lymphotoxin-β receptor. Molecular genetics, ligand binding, and expression. J Immunol 1995; 155:5280-8; PMID:7594541
- Croft M, Duan W, Choi H, Eun SY, Madireddi S, Mehta A. TNF superfamily in inflammatory disease: translating basic insights. Trends Immunol 2012; 33:144-52; PMID:22169337; http://dx.doi.org/10.1016/j.it.2011.10.004
- Fan K, Wang H, Wei H, Zhou Q, Kou G, Hou S, Qian W, Dai J, Li B, Zhang Y, et al. Blockade of LIGHT/HVEM and B7/CD28 signaling facilitates long-term islet graft survival with development of allospecific tolerance. Transplantation 2007; 84:746-54; PMID:17893608; http://dx.doi.org/10.1097/01.tp.0000280545.14489.df
- Ye Q, Fraser CC, Gao W, Wang L, Busfield SJ, Wang C, Qiu Y, Coyle AJ, Gutierrez-Ramos JC, Hancock WW. Modulation of LIGHT-HVEM costimulation prolongs cardiac allograft survival. J Exp Med 2002; 195:795-800; PMID:11901205; http://dx.doi.org/10.1084/jem.20012088
- Tamada K, Shimozaki K, Chapoval AI, Zhu G, Sica G, Flies D, Boone T, Hsu H, Fu YX, Nagata S, et al. Modulation of T-cell-mediated immunity in tumor and graft-vs.-host disease models through the LIGHT co-stimulatory pathway. Nat Med 2000; 6:283-9; PMID:10700230; http://dx.doi.org/10.1038/73136
- Scheu S, Alferink J, Potzel T, Barchet W, Kalinke U, Pfeffer K. Targeted disruption of LIGHT causes defects in costimulatory T cell activation and reveals cooperation with lymphotoxin β in mesenteric lymph node genesis. J ExpMed 2002; 195:1613-24; http://dx.doi.org/10.1084/jem.20020215
- Tamada K, Shimozaki K, Chapoval AI, Zhai Y, Su J, Chen SF, Hsieh SL, Nagata S, Ni J, Chen L. LIGHT, a TNF-like molecule, costimulates T cell proliferation and is required for dendritic cell-mediated allogeneic T cell response. J Immunol 2000; 164:4105-10; PMID:10754304; http://dx.doi.org/10.4049/jimmunol.164.8.4105
- Morel Y, Truneh A, Sweet RW, Olive D, Costello RT. The TNF superfamily members LIGHT and CD154 (CD40 ligand) costimulate induction of dendritic cell maturation and elicit specific CTL activity. J Immunol 2001; 167:2479-86; PMID:11509586; http://dx.doi.org/10.4049/jimmunol.167.5.2479
- Holmes TD, Wilson EB, Black EV, Benest AV, Vaz C, Tan B, Tanavde VM, Cook GP. Licensed human natural killer cells aid dendritic cell maturation via TNFSF14/LIGHT. Proc Natl Acad Sci U S A 2014; 111:E5688-96; PMID:25512551; http://dx.doi.org/10.1073/pnas.1411072112
- Wan X, Zhang J, Luo H, Shi G, Kapnik E, Kim S, Kanakaraj P, Wu J. A TNF family member LIGHT transduces costimulatory signals into human T cells. J Immunol 2002; 169:6813-21; PMID:12471113; http://dx.doi.org/10.4049/jimmunol.169.12.6813
- Shi G, Luo H, Wan X, Salcedo TW, Zhang J, Wu J. Mouse T cells receive costimulatory signals from LIGHT, a TNF family member. Blood 2002; 100:3279-86; PMID:12384428; http://dx.doi.org/10.1182/blood-2002-05-1404
- Granger SW, Rickert S. LIGHT-HVEM signaling and the regulation of T cell-mediated immunity. Cytokine Growth Factor Rev 2003; 14:289-96; PMID:12787566; http://dx.doi.org/10.1016/S1359-6101(03)00031-5
- Xu Y, Flies AS, Flies DB, Zhu G, Anand S, Flies SJ, Xu H, Anders RA, Hancock WW, Chen L, et al. Selective targeting of the LIGHT-HVEM costimulatory system for the treatment of graft-versus-host disease. Blood 2007; 109:4097-104; PMID:17179227; http://dx.doi.org/10.1182/blood-2006-09-047332
- Chattopadhyay PK, Yu J, Roederer M. A live-cell assay to detect antigen-specific CD4+ T cells with diverse cytokine profiles. Nat Med 2005; 11:1113-7; PMID:16186817; http://dx.doi.org/10.1038/nm1293
- Zhu M, Yang Y, Wang Y, Wang Z, Fu YX. LIGHT regulates inflamed draining lymph node hypertrophy. J Immunol 2011; 186:7156-63; PMID:21572030; http://dx.doi.org/10.4049/jimmunol.1002097
- Del Rio ML, Jones ND, Buhler L, Norris P, Shintani Y, Ware CF, Rodriguez-Barbosa JI. Selective blockade of herpesvirus entry mediator-B and T lymphocyte attenuator pathway ameliorates acute graft-versus-host reaction. J Immunol 2012; 188:4885-96; PMID:22490863; http://dx.doi.org/10.4049/jimmunol.1103698
- Fan Z, Yu P, Wang Y, Wang Y, Fu ML, Liu W, Sun Y, Fu YX. NK-cell activation by LIGHT triggers tumor-specific CD8+ T-cell immunity to reject established tumors. Blood 2006; 107:1342-51; PMID:16223768; http://dx.doi.org/10.1182/blood-2005-08-3485
- Pulaiev RA, Puliaeva IA, Ryan AE, Via CS. The Parent-into-F1 Model of Graft-vs-Host Disease as a Model of In Vivo T Cell Function and Immunomodulation. Curr Med Chem Immunol Endocr Metab Agents 2005; 5:575-83; PMID:19865585; http://dx.doi.org/10.2174/156801305774962204
- Tschetter JR, Mozes E, Shearer GM. Progression from acute to chronic disease in a murine parent-into-F1 model of graft-versus-host disease. J Immunol 2000; 165:5987-94; PMID:11067962; http://dx.doi.org/10.4049/jimmunol.165.10.5987
- Sarkar S, Kalia V, Haining WN, Konieczny BT, Subramaniam S, Ahmed R. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J Exp Med 2008; 205:625-40; PMID:18316415; http://dx.doi.org/10.1084/jem.20071641
- Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol 2003; 4:1191-8; PMID:14625547; http://dx.doi.org/10.1038/ni1009
- Ferrer IR, Wagener ME, Song M, Ford ML. CD154 blockade alters innate immune cell recruitment and programs alloreactive CD8+ T cells into KLRG-1(high) short-lived effector T cells. PloS One 2012; 7:e40559; PMID:22792369; http://dx.doi.org/10.1371/journal.pone.0040559
- Ware CF. Protein therapeutics targeted at the TNF superfamily. Adv Pharmacol (San Diego, Calif) 2013; 66:51-80; http://dx.doi.org/10.1016/B978-0-12-404717-4.00002-0
- Page EK, Dar WA, Knechtle SJ. Biologics in organ transplantation. TransplInt 2012; 25:707-19; http://dx.doi.org/10.1111/j.1432-2277.2012.01456.x
- Miller JF, Morahan G. Peripheral T cell tolerance. Annu Rev Immunol 1992; 10:51-69; PMID:1590995; http://dx.doi.org/10.1146/annurev.iy.10.040192.000411
- van Parijs L, Perez VL, Abbas AK. Mechanisms of peripheral T cell tolerance. Novartis Found Symp 1998; 215:5-14; PMID:9760568; http://dx.doi.org/10.1002/9780470515525.ch2
- Schwartz RH. T cell anergy. Annu Rev Immunol 2003; 21:305-34; PMID:12471050; http://dx.doi.org/10.1146/annurev.immunol.21.120601.141110
- Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature 1998; 393:480-3; PMID:9624005; http://dx.doi.org/10.1038/31002
- Judge TA, Wu Z, Zheng XG, Sharpe AH, Sayegh MH, Turka LA. The role of CD80, CD86, and CTLA4 in alloimmune responses and the induction of long-term allograft survival. J Immunol 1999; 162:1947-51; PMID:9973463
- Hargreaves RE, Monk NJ, Jurcevic S. Selective depletion of activated T cells: the CD40L-specific antibody experience. Trends Mol Med 2004; 10:130-5; PMID:15102356; http://dx.doi.org/10.1016/j.molmed.2004.01.009
- Tran HM, Nickerson PW, Restifo AC, Ivis-Woodward MA, Patel A, Allen RD, Strom TB, O'Connell PJ. Distinct mechanisms for the induction and maintenance of allograft tolerance with CTLA4-Fc treatment. J Immunol (Baltimore, Md : 1950) 1997; 159:2232-9
- Wekerle T, Grinyo JM. Belatacept: from rational design to clinical application. Transpl Int 2012; 25:139-50; PMID:22151353; http://dx.doi.org/10.1111/j.1432-2277.2011.01386.x
- Ford ML, Larsen CP. Translating costimulation blockade to the clinic: lessons learned from three pathways. Immunol Rev 2009; 229:294-306; PMID:19426229; http://dx.doi.org/10.1111/j.1600-065X.2009.00776.x
- Williams MA, Trambley J, Ha J, Adams AB, Durham MM, Rees P, Cowan SR, Pearson TC, Larsen CP. Genetic characterization of strain differences in the ability to mediate CD40/CD28-independent rejection of skin allografts. J Immunol 2000; 165:6849-57; PMID:11120808; http://dx.doi.org/10.4049/jimmunol.165.12.6849
- Trambley J, Bingaman AW, Lin A, Elwood ET, Waitze SY, Ha J, Durham MM, Corbascio M, Cowan SR, Pearson TC, et al. Asialo GM1(+) CD8(+) T cells play a critical role in costimulation blockade-resistant allograft rejection. JClinInvest 1999; 104:1715-22; http://dx.doi.org/10.1172/JCI8082
- Doherty TA, Croft M. Therapeutic potential of targeting TNF/TNFR family members in asthma. Immunotherapy 2011; 3:919-21; PMID:21843077; http://dx.doi.org/10.2217/imt.11.88
- Cheung TC. Modulation of T cell proliferation through the LIGHT-HVEM-BTLA cosignaling pathway. Recent Pat DNA Gene Seq 2009; 3:177-82; PMID:19702559; http://dx.doi.org/10.2174/187221509789318342
- Gommerman JL, Browning JL. Effective expression and purification of bioactive recombinant soluble LIGHT. Nat Rev Immunol 2003; 3:642-55; PMID:12974479; http://dx.doi.org/10.1038/nri1151
- Tsuji I, Iwamoto K, Shintani Y. Effective expression and purification of bioactive recombinant soluble LIGHT. Methods Mol Biol (Clifton, NJ) 2014; 1155:201-13; http://dx.doi.org/10.1007/978-1-4939-0669-7_17
- Wang J, Lo JC, Foster A, Yu P, Chen HM, Wang Y, Tamada K, Chen L, Fu YX. The regulation of T cell homeostasis and autoimmunity by T cell-derived LIGHT. J Clin Invest 2001; 108:1771-80; PMID:11748260; http://dx.doi.org/10.1172/JCI200113827
- Anand S, Wang P, Yoshimura K, Choi IH, Hilliard A, Chen YH, Wang CR, Schulick R, Flies AS, Flies DB, et al. Essential role of TNF family molecule LIGHT as a cytokine in the pathogenesis of hepatitis. J Clin Invest 2006; 116:1045-51; PMID:16557300; http://dx.doi.org/10.1172/JCI27083
- Fava RA, Notidis E, Hunt J, Szanya V, Ratcliffe N, Ngam-Ek A, De Fougerolles AR, Sprague A, Browning JL. A role for the lymphotoxin/LIGHT axis in the pathogenesis of murine collagen-induced arthritis. J Immunol (Baltimore, Md : 1950) 2003; 171:115-26; http://dx.doi.org/10.4049/jimmunol.171.1.115
- Herro R, Da Silva Antunes R, Aguilera AR, Tamada K, Croft M. Tumor necrosis factor superfamily 14 (LIGHT) controls thymic stromal lymphopoietin to drive pulmonary fibrosis. J Allergy Clin Immunol 2015; 136(3):757-68; PMID:25680454
- Doherty TA, Soroosh P, Khorram N, Fukuyama S, Rosenthal P, Cho JY, Norris PS, Choi H, Scheu S, Pfeffer K, et al. The tumor necrosis factor family member LIGHT is a target for asthmatic airway remodeling. Nat Med 2011; 17:596-603; PMID:21499267; http://dx.doi.org/10.1038/nm.2356
- Herro R, Antunes Rda S, Aguilera AR, Tamada K, Croft M. The Tumor Necrosis Factor Superfamily Molecule LIGHT Promotes Keratinocyte Activity and Skin Fibrosis. J Invest Dermatol 2015; 135:2109-18; PMID:25789702; http://dx.doi.org/10.1038/jid.2015.110
- Brunetti G, Rizzi R, Oranger A, Gigante I, Mori G, Taurino G, Mongelli T, Colaianni G, Di Benedetto A, Tamma R, et al. LIGHT/TNFSF14 increases osteoclastogenesis and decreases osteoblastogenesis in multiple myeloma-bone disease. Oncotarget 2014; 5:12950-67; PMID:25460501; http://dx.doi.org/10.18632/oncotarget.2633
- Bruhns P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood 2012; 119:5640-9; PMID:22535666; http://dx.doi.org/10.1182/blood-2012-01-380121
- Bournazos S, DiLillo DJ, Ravetch JV. The role of Fc-FcgammaR interactions in IgG-mediated microbial neutralization. J Exp Med 2015; PMID:26282878; http://dx.doi.org/10.1084/jem.20151267
- Nimmerjahn F, Ravetch JV. Divergent immunoglobulin g subclass activity through selective Fc receptor binding. Science (New York, NY) 2005; 310:1510-2; http://dx.doi.org/10.1126/science.1118948
- Biburger M, Lux A, Nimmerjahn F. How immunoglobulin G antibodies kill target cells: revisiting an old paradigm. Adv Immunol 2014; 124:67-94; PMID:25175773; http://dx.doi.org/10.1016/B978-0-12-800147-9.00003-0
- Cudkowicz G, Bennett M. Peculiar immunobiology of bone marrow allografts. II. Rejection of parental grafts by resistant F 1 hybrid mice. J Exp Med 1971; 134:1513-28; PMID:4942407; http://dx.doi.org/10.1084/jem.134.6.1513
- Soloviova K, Puliaiev M, Foster A, Via CS. The parent-into-F1 murine model in the study of lupus-like autoimmunity and CD8 cytotoxic T lymphocyte function. Methods Mol Biol (Clifton, NJ) 2012; 900:253-70; http://dx.doi.org/10.1007/978-1-60761-720-4_12
- Summers-DeLuca LE, McCarthy DD, Cosovic B, Ward LA, Lo CC, Scheu S, Pfeffer K, Gommerman JL. Expression of lymphotoxin-alphabeta on antigen-specific T cells is required for DC function. J Exp Med 2007; 204:1071-81; PMID:17452522; http://dx.doi.org/10.1084/jem.20061968
- Gommerman JL, Summers dL. LTbetaR and CD40: working together in dendritic cells to optimize immune responses. Immunol Rev 2011; 244:85-98; PMID:22017433; http://dx.doi.org/10.1111/j.1600-065X.2011.01056.x
- Del Rio ML, Schneider P, Fernandez-Renedo C, Perez-Simon JA, Rodriguez-Barbosa JI. LIGHT/HVEM/LTbetaR Interaction as a Target for the Modulation of the Allogeneic Immune Response in Transplantation. Am J Transplant 2013; 13:541-51; PMID:23356438; http://dx.doi.org/10.1111/ajt.12089
- Schenk AD, Gorbacheva V, Rabant M, Fairchild RL, Valujskikh A. Effector functions of donor-reactive CD8 memory T cells are dependent on ICOS induced during division in cardiac grafts. Am J Transplantation 2009; 9:64-73; http://dx.doi.org/10.1111/j.1600-6143.2008.02460.x
- Rodriguez-Barbosa JI, Gutierrez Martin CB, Tascon RI, Suarez J, Rodriguez Ferri EF. Evidence obtained with monoclonal antibodies that O antigen is the major antigen responsible for the cross-reactivities between serotypes 4 and 7 of Actinobacillus (Haemophilus) pleuropneumoniae. Clin Diagn Lab Immunol 1995; 2:563-8; PMID:8548535
- Ito T, Iwamoto K, Tsuji I, Tsubouchi H, Omae H, Sato T, Ohba H, Kurokawa T, Taniyama Y, Shintani Y. Trimerization of murine TNF ligand family member LIGHT increases the cytotoxic activity against the FM3A mammary carcinoma cell line. Appl Microbiol Biotechnol 2011; 90:1691-9; PMID:21400099; http://dx.doi.org/10.1007/s00253-011-3168-8
- Schneider P, Willen L, Smulski CR. Tools and techniques to study ligand-receptor interactions and receptor activation by TNF superfamily members. Meth Enzymol 2014; 545:103-25; PMID:25065888; http://dx.doi.org/10.1016/B978-0-12-801430-1.00005-6
- Nguyen HH, Park J, Kang S, Kim M. Surface plasmon resonance: a versatile technique for biosensor applications. Sensors (Basel, Switzerland) 2015; 15:10481-510; PMID:25951336; http://dx.doi.org/10.3390/s150510481
- Del Rio ML, Kaye J, Rodriguez-Barbosa JI. Detection of protein on BTLA(low) cells and in vivo antibody-mediated down-modulation of BTLA on lymphoid and myeloid cells of C57BL/6 and BALB/c BTLA allelic variants. Immunobiology 2010; 215:570-8; http://dx.doi.org/10.1016/j.imbio.2009.09.008
- Unkeless JC. Characterization of a monoclonal antibody directed against mouse macrophage and lymphocyte Fc receptors. J Exp Med 1979; 150:580-96; PMID:90108; http://dx.doi.org/10.1084/jem.150.3.580
- Lyons AB. Analysing cell division in vivo and in vitro using flow cytometric measurement of CFSE dye dilution. J Immunol Methods 2000; 243:147-54; PMID:10986412; http://dx.doi.org/10.1016/S0022-1759(00)00231-3