
ABSTRACT
Herein we describe the investigation of a Chinese hamster ovary (CHO)-expressed human mAb molecule found partially modified by a +80 Da adduct. This mass difference, suggestive of a single sulfation or phosphorylation addition, was observed by mass analysis of the intact and reduced molecule by mass spectrometry (MS). The modification was located on tyrosine 31 (Y31) of the light chain in the complementarity-determining region 1 by liquid chromatography (LC)-MS peptide mapping and electron transfer dissociation fragmentation. The complete loss of the 80 Da modification moiety during collision induced dissociation fragmentation suggested this modification could not be a tyrosine phosphorylation. Treatment of the mAb with alkaline phosphatase confirmed our hypothesis. Western blot experiment using anti-tyrosine sulfation antibody and LC retention time correlation with corresponding synthetic sulfated peptides further confirmed the identification of tyrosine sulfation on the light chain. The unique sequence motif with neighboring acidic amino acids and local secondary structure might play a role to make Y31 a substrate residue for sulfation. This type of modification, to our knowledge, has not been previously reported for CHO-produced human IgG antibodies.
Introduction
Tyrosine sulfation is a post-translational modification (PTM) in which a sulfate trioxide (SO3) group is covalently bound to the hydroxyl group on the side chain of the amino acid tyrosine group. This PTM occurs in the trans-Golgi network and is catalyzed by 2 enzymes, tyrosylprotein sulfotransferases (TPSTs) 1 and 2. The molecular mechanism involves the transfer of an activated sulfate from 3′-phosphoadenosine-5′-phsophosulfate to tyrosine, and has been found on a variety of proteins and peptides.Citation1,2 Recent findings indicate that tyrosylprotein sulfotransferase 2 recognizes tyrosines flanked by acid residues for sulfation.Citation3 This PTM is responsible for strengthening interactions between proteins and occurs almost exclusively on secreted and trans-membrane spanning proteins.Citation4-11 Many chemokine receptors have been shown to be tyrosine sulfated such as N-terminal extracellular domain of CCR5, the principle HIV-1 and several glycoprotein hormone receptors.Citation6,12,13 For example, the native form of the leech-derived thrombin inhibiting peptide hirudin is tyrosine sulfated. Interestingly, the 2 recombinant forms of hirudin (Revasc and Refludan) used for treating various blood clotting disorders are not sulfated. Subsequent studies have shown that sulfated hirudin displays a tenfold higher affinity for thrombin than the un-sulfated analogs, thus illustrating tyrosine sulfation's biologic relevance.Citation13-15 Sulfation increases the mass of a biomolecule by 80 Da, which is the same mass difference as a phosphate moiety (PO3). Unlike PO3, which forms a fairly stable P-O bond, the SO3 is very labile and readily decomposes under high temperature and low pH conditions. This makes it a challenge to analyze via many traditional analytical tools. In-depth characterization of this post-translational modification has been hampered by the lack of general, unambiguous methods for site determination, because of the labile nature of sulfotyrosine.
Tyrosine sulfation occurs in higher eukaryotic species and in all mammalian cell lines tested thus far, but not in prokaryotes or in yeast. Chinese hamster ovary (CHO) cells have become the major expression system for the manufacture of human therapeutic proteins. To our knowledge, no CHO-derived monoclonal antibodies (mAbs) have been reported with tyrosine sulfation. Herein we describe the investigation of a CHO-expressed human mAb molecule found partially modified by a +80 Da adduct. This mass difference suggests the post-translational addition of a sulfate molecule. The location of this labile modification was found in complementarity-determining region (CDR) 1 of the light chain, and was identified by mass spectrometry with electron transfer dissociation (ETD) fragmentation. No response to treatment of alkaline phosphatase provides strong evidence against the possibility of tyrosine phosphorylation. To confirm the tyrosine sulfation, western blotting experiments using an anti-tyrosine sulfation antibody and retention time correlation with synthetic sulfated peptide was performed. Additionally, the acidic amino acid residues near to the targeted tyrosine and elements of local secondary structure might play an essential role to make this modification happen.
Results
Separation of mAb molecule
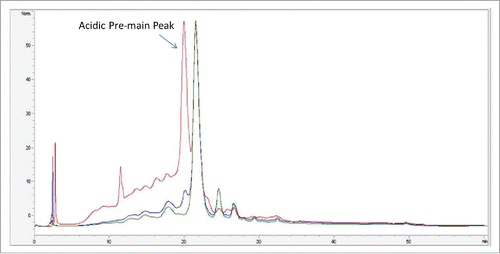
Anion exchange chromatography (AEX) is typically used as a polishing step during mAb purification. This step typically is operated in a flow through mode, where the mAb flows through the column and in-process impurities (host cell proteins, DNA) bind to the column. During the AEX development, it was noted that a fraction of the mAb loaded on the column bound to the resin, which affects protein recovery. The bound fraction of the protein eluted in the strip fraction of the AEX chromatography. To characterize the mAb bound to AEX column, fractions of the AEX chromatography were analyzed: “load” refer to the sample before AEX purification; “pool” refers to flow through portion of the sample and “strip” refers to the bound fraction of the sample. Load, pool and strip fractions from AEX chromatography were initially analyzed by strong cation exchange (SCX)-HPLC chromatography. shows the normalized SCX-HPLC profile of mAb in the AEX load, pool and strip fractions. As shown in , the strip fraction had a significantly higher amount of acidic variants compared with the load and pool: ∼65% acid variants in the strip fraction (red trace) compared with 23% in the pool fraction (green trace) and 33% in the load fraction (blue trace). An additional difference was noted in the acidic pre-main peak. This peak was present in the AEX load fraction at higher levels, whereas in the AEX pool, this peak was minimal. The strip fraction was enriched with the acidic pre-main peak.
Figure 1. Overlay of SCX-HPLC UV profile of AEX load (blue trace), strip (red trace) and pool fraction (green trace).
Analysis of intact and reduced protein by mass spectrometry
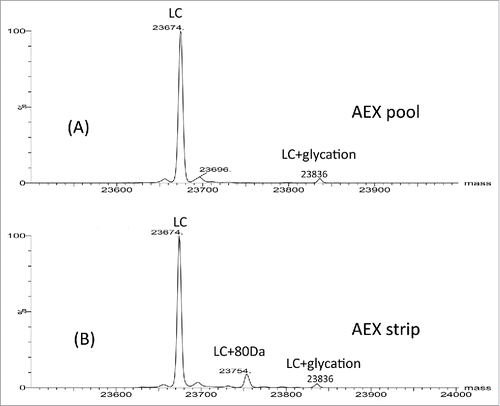
To characterize the impurities, all 3 fractions (AEX load, pool and strip) were analyzed by intact and reduced LC/MS using Q-TOF MS. shows the deconvoluted mass spectra of the intact molecule. Three main glycoforms were observed in all 3 fractions: G0F/G0F, G0F/G1F and G1F/G1F with mass of 148591 Da, 148751 Da and 148912 Da, respectively. The calculated intact mass of this molecule with G0F/G0F is 148590 Da. The mass errors for intact mass measurement are all within 25 ppm. Additional species were only detected in the AEX strip fraction. These species correspond to 80 Da mass increase (148668, 148830, 148991 Da) of the 3 major glycoforms (G0F/G0F, G1F/G0F, G1F/G1F). Based on the intensity of deconvoluted intact mass in , ∼40% of the proteins in the strip fraction are modified with 80 Da addition. To locate the modification, the light chain and heavy chain mass were measured after the disulfide bonds cleavage by the reducing agent dithiothreitol (DTT). No difference was detected on heavy chain mass of the strip fraction and pool fraction, suggesting the modification is not located on heavy chain (data not shown). As shown in and , the light chain apo form mass (23674 Da) and glycated light chain mass (23836 Da) were detected in both fractions (strip and pool fraction). A peak with 80 Da increase of light chain was only observed at 23754 Da in the AEX strip fraction. The mass error of reduced mass measurement is within 20 ppm. These data suggest that the 80 Da modifications are located on light chain of the mAb.
Figure 2. Intact mass spectra of (A) AEX load, (B) pool and (C) strip samples.
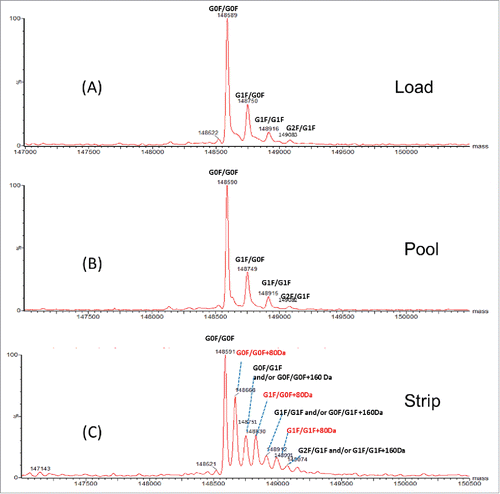
Figure 3. Reduced light chain mass spectra of AEX (A) pool and (B) strip samples.
Analysis of mAb by reduced peptide mapping
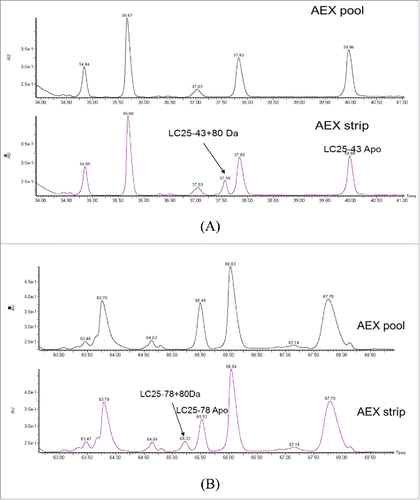
To further locate the modification site, the AEX strip and pool fractions were reduced, alkylated and then digested by LysC enzyme. The peptide mixtures were mass mapped by Q-TOF MS. When comparing the UV trace of these 2 fractions, 2 differences were noticed. As shown in and , 2 new peaks were detected at retention time 37.6 min and 65.2 min in the AEX strip fraction. The observed m/z in the new peaks are 1165.4796 (2+) at 37.6 min and 1476.7372 (4+) Da at 65.2 min. The observed masses correspond to light chain peptide AA25–43+80 Da and AA25–78 +80 Da with mass error of 6.4 ppm and 9.8 ppm, respectively. Light chain peptide AA25–78 contains one miscleavage site. The modified and unmodified form of light chain peptide AA25–43 and AA25–78 were labeled in . The level of this modified peptide was estimated to be 20.9% and 21.6% for AA25–43 and AA25–78, respectively, compared with their apo forms based on the peak area in extracted ion chromatogram (SIC).
Figure 4. UV chromatograms of reduced LysC peptide mapping of AEX (A) pool and (B) strip fractions.
MS/MS fragmentation of the modified peptide
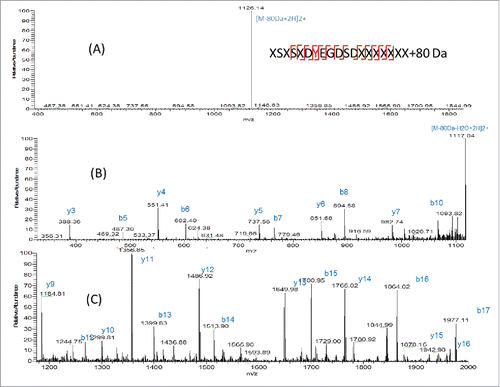
There are 2 possibilities of modification with 80 Da increase in mass: phosphorylation (+79.9663 Da) and sulfation (+79.9568 Da). The theoretical mass difference of these 2 modifications is only 0.0095 Da. The measured m/z of AA25–43+80 Da peptide is 1165.4645 (2+) using Orbitrap MS. The mass error is 0.3 ppm compared with the theoretical mass of this peptide with sulfation (1165.4648, 2+) and 4.4 ppm compared with this peptide with phosphorylation (1165.4696, 2+). Although the mass error is smaller for sulfation, it is still difficult to distinguish sulfation and phosphorylation by mass only. Further experiment needs to be performed for identification. Initially, the target peptide AA25–78 was fragmented by collision-induced dissociation (CID) and the produced fragments were analyzed in the linear ion trap of the LTQ-Orbitrap MS. As shown in , a dominant precursor ion with complete loss of the modification group (80 Da) was observed. Only fragments from the peptide backbone were detected, as shown in the zoomed-in spectra in (m/z 300–1100) and (m/z 1200–2000). A series of b (b5-b8, b10, b12-b17) and y ions (y3-y7 y9-y16) from peptide backbone fragmentation were detected, which confirms the light chain peptide sequence of AA25–43. No site-specific information was obtained from CID fragmentation. It has been reported that sulfated tyrosine (sY) is very labile and could be easily lost under standard CID conditions.Citation16 It was not possible to obtain site-specific information of the sulfation modification using the positive ion CID MS/MS, as none of the original precursor ions with the 80 Da adduct were present at the time of peptide backbone fragmentation and no diagnostic ion of tyrosine sulfation was detected. In contrast, phosphorylated peptides tend to persist under CID and peptide backbone fragmentation allows for the site-specific identification of the modification.Citation16 In , a neutral loss of 80 Da from precursor ion was observed. It is known that the characteristic neutral loss ion for phosphorylation is -H3PO4 (−98 Da) and characteristic fragment ion is PO3− (−79 Da), while for sulfation, the characteristic neutral loss ions and fragment ions are both -SO3 ion with 80 Da.Citation3 The CID MS2 data suggests the 80 Da modification is sulfation.
Figure 5. (A) CID fragmentation spectrum of light chain AA25–43+80 Da in 400–1800 m/z (B) Zoom in m/z 300–1100 (C) Zoomed in m/z 1200–2000.
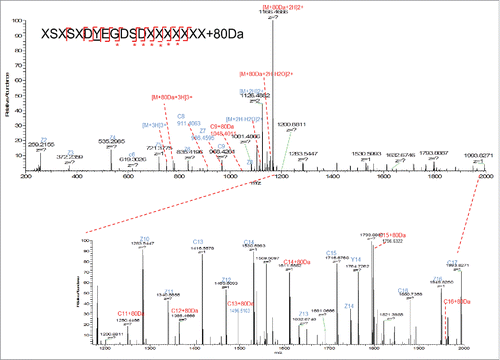
Another widely used fragmentation mechanism is ETD. It transfers electrons to a multiply protonated peptide/protein, which could lead to the cleavage of the N-Cα backbone bonds and generate c- and z-type fragment ions without loss of the information of the PTM localization.Citation17 ETD can provide complementary information with CID. The ETD process allows retention of the SO3 group and thus the amino acid localization, while CID preferably fragments labile modifications. In our case, the target peptide was analyzed by LTQ-Orbitrap with ETD fragmentation and high-resolution mass detection in Orbitrap MS. As shown in , partial loss of 80 Da modifications was observed on the precursor ion. Fragment ions with SO3 group (+80 Da) attached are labeled in red. Based on the detection of SO3 attached fragment ions (c9, c11, c12–16) and the fact that there is only one tyrosine in this peptide, the modification site was identified to be tyrosine 31 on light chain, which is in the CDR1 region of the mAb molecule. This modification site was further confirmed by comparison with the sulfated synthetic peptide with the same sequence (data shown in later section).
Figure 6. ETD fragmentation of light chain peptide AA25–43+80 Da. The 80 Da attached fragment ions were highlighted in red and labeled with *.
Alkaline phosphatase treatment
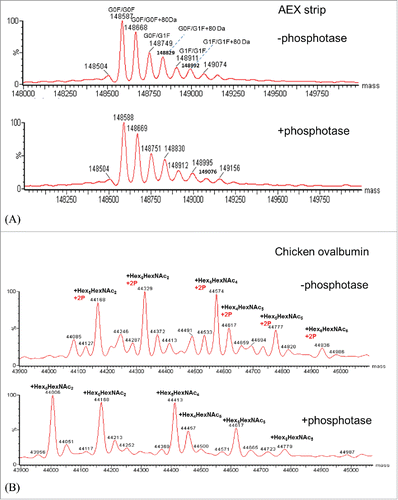
Since phosphorylation and sulfation of tyrosine are isobaric and directly distinguishing between tyrosine-sulfation and phosphorylation could be challenging, alkaline phosphatase was also used to distinguish these 2 modifications.Citation18 Alkaline phosphatase has been widely used for removing phosphorylation group from proteins. Chicken albumin was used as a positive control as this protein has been widely known for its phosphorylation and glycosylation forms. Chicken albumin and mAb in the AEX strip fraction were treated with phosphatase and incubate at 37°C side by side. shows the measured intact mass of the mAb and chicken ovalbumin before and after phosphatase treatment. As shown in , no mass change was observed for the mAb, while for chicken albumin () an obvious mass shift of 160 Da was observed for all the major glycoforms. Since chicken albumin contains 2 phosphorylation sites, the loss of 160 Da confirms the activity of alkaline phosphatase. As no mass change was detected before and after phosphatase treatment, it suggests that this mAb in the AEX strip fraction is not phosphorylated. Thus far, LC/MS analysis has been used to investigate the nature of the 80 Da adduct in the light chain CDR1. MS2 analysis and mass analysis of the mAb AEX strip fraction after phosphatase-treatment had suggested that the 80 Da adduct is sulfation on tyrosine 31.
Figure 7. (A) Deconvoluted intact mass spectra of AEX strip fraction with and without alkaline phosphatase treatment. (B) Deconvoluted intact mass spectra of chicken ovalbumin with and without alkaline phosphatase treatment.
Western blot
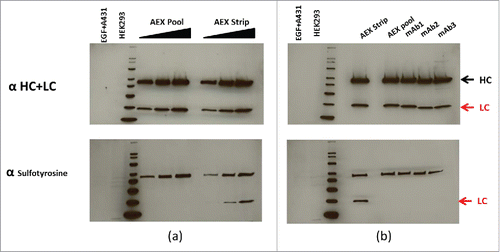
In a continued effort to confirm the identification of sulfation of tyrosine, western blotting with an anti-sulfotyrosine-specific mAb was applied to confirm the presence of tyrosine sulfation in the AEX strip fraction of the mAb.Citation19 In (upper panel), the normalized concentrations of mAb AEX pool and strip fractions were subjected to reduced SDS-PAGE, probed for the human heavy and light chains by western hybridization. The increased concentrations of heavy and light chains from the pool and strip fractions were then “stripped” of the first detecting antibodies and re-probed with an anti-sulfotyrosine-specific monoclonal antibody (lower panel). As shown in (lower panel), positive signals were detected on the light chain of the strip fraction of the mAb, suggesting that it contains tyrosine sulfation. Positive response was also observed on the heavy chains. This is due to non-specific detection, since no molecular weight increase corresponding to sulfation was observed on the heavy chain by both reduced mass and peptide mass analysis. Furthermore, this positive response was detected on all the different mAbs we tested. It has been shown in the past that denatured heavy and light chains can bind detection antibodies non-specifically.Citation20 As a control for cross reactivity with phosphorylation, lane 1 was loaded with a commercial source of EGF-treated A431 cell extract that is enriched with phosphorylated proteins. The lack of positive signal in lane 1 of the lower panel shows the anti-sulfotyrosine mAb does not have strong cross reactivity with phosphorylation (, lower panel). Further, HEK 293 extract, which was suggested by the manufacturer as a positive control for the anti-sulfotyrosine mAb, was loaded in lane 2. The positive signal below the bottom 20 kDa is consistent with the manufacturer's analysis (, lower panel). In , normalized concentrations of different CHO-derived mAbs (mAb1, 2 and 3) in addition to the AEX strip and pool are subjected to reduced SDS-PAGE, probed for the human heavy and light chains by western hybridization (upper panel), then stripped and re-probed for anti-sulfotyrosine (lower panel). In agreement with , the AEX mAb strip shows a positive signal at the light chain position when probed with the anti-sulfotyrosine antibody. No positive signal was observed on tyrosine sulfation for the light chains of the other 3 CHO-derived Merck mAbs when similar amounts of protein were analyzed. This is consistent with our observation that no increased level of AEX acidic peak or tyrosine sulfation hotspot was detected on these 3 mAbs (data not shown). Again, non-specific detection was observed on the heavy chains of all 3 mAbs tested.
Figure 8. (a) Normalized concentrations of mAb AEX pool and strip were subjected to reduced SDS-PAGE, probed for the human heavy (HC) and light chains (LC) by western hybridization (upper panel), then stripped and re-probed for antisulfotyrosine (lower panel). See the indications for HC and LC at the far right. (b) Normalized concentrations of different CHO-derived mAbs in addition to AEX strip and pool are subjected to reduced SDS PAGE, probed for the human HC and LC by western hybridization, then stripped and re-probed for anti sulfotyrosine. For both (a) and (b) MagicMark XP was used as a protein molecular weight standard, and equal amounts of HEK293 and EGF-treated A431 cell extracts are analyzed as controls.
Comparison of liquid chromatography retention time with synthetic peptides modified by sulfation or phosphorylation
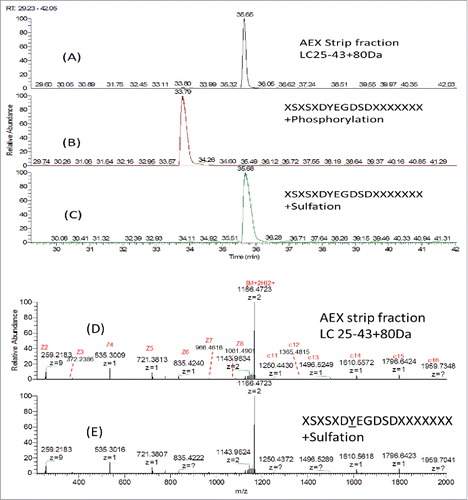
To further distinguish phosphorylation and sulfation, synthetic peptide with identical sequence of LC AA25–43 (XSXSXDYEGDSDXXXXXXX) modified with either phosphorylation or sulfation on the Y31 were analyzed by LC/MS. shows the SIC of AA25–43+80 Da in the AEX strip fraction, synthetic peptide XSXSXDpYEGDSDXXXXXXX and XSXSXDsYEGDSDXXXXXXX. Synthetic peptide with sulfation elutes at the same retention time as the peptide AA25–43+80 Da detected in the AEX strip fraction, while the synthetic peptide with phosphorylation elutes earlier than that detected in the AEX strip. The ETD MS2 spectra of the synthetic peptide with sulfation and the peptide AA25–43+80 Da in the AEX strip fraction were shown in and . In addition to the same retention time being observed, the almost identical MS2 profile suggested that these 2 peptides have the same amino acid sequence and site of modification. This further confirms our observation that Y31 on light chain is sulfated.
Figure 9. SIC of (A) LC25–43C80 Da from AEX Strip Fraction, (B) Synthetic peptide XSXSXDYEGDSDXXXXXXXCPhosphorylation and (C) Synthetic peptide XSXSXDYEGDSDXXXXXXXCSulfation; ETD MS2 spectra of (D) LC25–43C80 Da from AEX strip fraction and (E) Synthetic peptide XSXSXDYEGDSDXXXXXXXCSulfation.
Structure modeling of tyrosine sulfation site
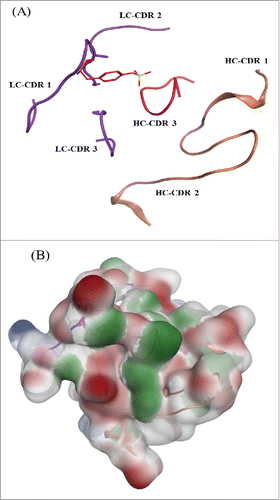
The protein tyrosine sulfation reaction is catalyzed by the Golgi enzyme tyrosylprotein sulfotransferase. Previous studies indicated that TPSTs recognize accessible tyrosine residues that are usually surrounded by several acidic residues within −5 to +5 positions.Citation21-23 The acceptor tyrosine needs to have acidic residues nearby to enable the recognition of positively charged residues in the TPST2 substrate binding site. The acceptor tyrosine also needs to be in an intrinsically flexible region to fit into the deep cleft of TPST2. However, no general consensus sequence for tyrosine sulfation sites has been defined. The most common features describing the sequence surroundings of sulfated tyrosine includes the presence of one acidic amino acid within 2 residues of the tyrosine; presence of at least 3 acidic amino acid within 5 residues and presence of turn-inducing amino acids nearby, etc.Citation3 shows the structure of the mAb tyrosine site in the context of CDR loops in ribbon diagram, which was generated by MOE software (Chemical Computing Group, Montreal, Canada). The sequence near to light chain Y31 on this mAb is: XSXSXDYEGDSDXXXXXXX. In this sequence, the adjacent residues of Y31 are both acidic: aspartic acid (D) and glutamic Acid (E). A total of 4 acidic residues are within 5 residues of Y31: 3 D and one E. Four turn-inducing residues are close to Y31: 3 serine (S) and one glycine (G). Further, the sequence motif alone does not guarantee the occurrence of this modification. Factors such as percent exposure of these residues and the acidic nature of the region surrounding the tyrosine in the folded state can influence the sulfation event. In addition, if the sulfation occurs post-folding “native state,” the primary sequence alone cannot guarantee sulfation. illustrates that the acidic nature of the flaking residues are conserved in the final fold and not disrupted by the nearby residues. The final structure retains a significant acidic region, which is consistent with the trends that have been previously reported.Citation3 The unique structure of Y31 with neighboring acidic amino acids and elements of local secondary structure might play a role to make this modification happen.
Figure 10. mAb tyrosine (Y31) site showing the CDR loops for both the heavy and light chain. (A) Sulfated tyrosine (Try31) is depicted in red sticks and in the context of the CDR regions in ribbon diagram. (B) Surface map of the CDR region shown in the same orientation as the ribbon diagram. Hydrophobic regions in green, negatively charges regions in red, and positively charged region in blue.
Discussion
We describe here the evidence that points to the presence of an unexpected O-linked tyrosine sulfation in a CHO-produced monoclonal antibody. Protein recovery of the AEX purification step was affected as this modification makes the protein bind to the AEX column. Higher acidic pre-main peak level was also observed on SCX profile of the modified mAb. The AEX strip fraction of the mAb enriched with sulfation was collected and subjected to a series of analysis for characterization.
The location of this labile modification was found in CDR1 region of light chain, as identified by reduced mass measurement and reduced LysC peptide mapping analysis. The modification site was identified to be Y31 by LC-MS/MS analysis with ETD fragmentation. No response to alkaline phosphatase suggests this mAb is not phosphorylated even though sulfation and phosphorylation cause almost the same mass changes to the mAb (+80 Da). This tyrosine sulfation was further confirmed by a western blot experiment using anti-tyrosine sulfation antibody and retention time correlation with synthetic sulfated peptide.
Although there is no clear-cut acceptor motif can be defined that allows the prediction of tyrosine sulfation sites in polypeptide chains, the sulfated tyrosine residues could potentially be recognized depending on their location within the sequence. The “Sulfinator” (http://web.expasy.org/sulfinator/) is a software tool that can be used to predict tyrosine sulfation sites in protein sequences with an overall accuracy of 98%.Citation24 The mAb sequence was searched using this software. Only Y31 shows a positive hit. No other tyrosine residues in this mAb share similar local sequence characteristics that are typical for sulfation. Structural analysis of CDR tyrosine confirms the effect of acidic residues and local structure on tyrosine sulfation. The neighboring acidic amino acid residues and elements of local secondary structure might play a role to make Y31 a hotspot for sulfation.
Materials and methods
MAbs were manufactured at Merck. Alkaline phosphatase was purchased from New England Biolabs (Cat# M0290S, Ipswich, MA). Anti-tyrosine sulfation antibody was purchased from Millipore (Cat# 05–1100, Billerica, MA). Synthetic peptides were purchased from AnaSpec (sulfated peptide: Cat#60362–2, lot# 1556817; phosphorylated peptide: Cat#60362–1, lot# 1556816, Fremont, CA).
Anion exchange chromatography
AEX chromatography was performed using POROS® GoPure™ D Pre-packed Column, 0.5 × 5 cm, 1 mL in a flow through mode by using a GE Akta Avant system. The protein A chromatography purified mAb was pH adjusted to pH 6.5 with 1 M Tris and was loaded on the column. Prior to protein loading, the column was equilibrated with 25 mM sodium phosphate pH 6.5, post loading the column was washed with 25 mM sodium phosphate pH 6.5 and striped with 1 M NaCl. The absorbance at 280 nm was monitored for the duration of the run. Fractions, pool and strip, and AEX load were collected and analyzed.
Ion exchange HPLC
Ion exchange HPLC was performed on a MabPac SCX-10 column (4 × 250 mm, 3.14 ml) at ambient temperature by using an Agilent 1600 series system. Mobile phase B was 30 mM sodium phosphate pH 8.0 and mobile A was 25 mM MES, pH 5.8. The column was first equilibrated at 14% mobile phase B at a flow rate of 1.0 mL/min for 10 min. The mAb protein was then eluted from the column using a gradient of mobile phase B (14% to 80% in 18 min). The column was then cleaned with 100% mobile B for 3 min and re-equilibrated at 14% mobile phase B for the next sample analysis. The absorbance at 280 nm of the eluate was monitored throughout the LC run.
Intact and reduced LC/MS
Twenty μg of a sample was diluted to 0.5 mg/mL with 50 mM Tris buffer pH 8.0. The RP-HPLC separation was performed using Waters Acquity UPLC H-class. The column used in this study was Acquity UPLC BEH300 C4, 1.7 μm, 1.0 × 100 mm (Waters, Milford, MA). 2 µg (4µL) samples were injected to the column. Mobile phases were 0.1% formic acid (FA) in water as mobile A and 0.1% FA in acetonitrile (ACN) as mobile B. The LC flow rate was 0.08 mL/min and the column temperature was maintained at 80°C. The antibody was eluted using a gradient of 4 – 15 min of 30% – 90% B. MS spectra were acquired on a Waters Xevo G2 Q-TOF system which was scanned in a range of m/z 800 – 4000.
Twenty μg of a sample was diluted by a reducing buffer (50 mM Tris pH 8.0, containing 6 M guanidine HCl) to a final volume of 100 μL. Two μL of 1 M DTT (Sigma-Aldrich, St. Louis, MO) solution was added to each of the samples followed by incubation at 56°C for 20 min. The RP-UPLC separation was performed on a Waters Acquity UPLC H-class. The column used was Acquity UPLC, BEH300 C4, 2.1 × 100 mm, 1.7 um (Waters). 2 µg reduced samples (10µL) were loaded to the column. MS spectra were acquired on a Waters Xevo G2 Q-TOF system which was scanned in a range of m/z 600 – 3000. MS data was analyzed by MaxEnt1 of MassLynx 4.1.
Peptide mapping LC/MS
100 μg of a sample was buffer exchanged to 100 uL denaturing buffer containing 50 mM Tris pH 8.0, 6 M Guanidine HCl and 5 mM EDTA. The reducing reactions were conducted at 56°C for 30 min with 20 mM DTT in the solution. The samples were alkylated with 50 mM iodoacetamide at room temperature for 30 min in dark. The alkylation reaction was terminated by adding 1μL of a 500 mM DTT solution. The reduced and alkylated samples were diluted with a digestion buffer (50 mM Tris pH 8.0) to a final volume of 300 μL, before adding Lys-C enzyme (Wako, Richmond, VA) with an enzyme:substrate ratio of 1:20 (w:w). The solution was incubated at 37°C for 4 hour. The peptides were separated by RP-HPLC on a Waters Acquity UPLC H-class using a HALO Peptide ES-C18, 2.1 × 150 nm, 2.7 μm column (MAC-MOD Analytical, Inc., Chadds Ford, PA). Mobile phases were 0.1% TFA in H2O as mobile phase A and 0.1% TFA in ACN as mobile phase B. The LC flow rate was 0.2 mL/min and the column temperature was maintained at 60°C. The LC gradient was 2 – 30 min 2% – 18% B, 30 – 90 min 18% – 40% B, and 90 – 100 min 40% – 45% B. MS spectra were acquired on a Waters Xevo G2 Q-TOF system scanned in a range of m/z 100 – 2000. MS data was analyzed by BiopharmaLynx 1.3 (Waters).
Target MS/MS
LC/MS/MS of target peptide was conducted on a LTQ-Orbitrap Velos MS system with ETD (Thermo Fisher, Waltham, MA). Resolution of 17500 in FT mode was applied for MS/MS acquisition. The peptides were separated by Waters Acquity UPLC H-class using a HALO Peptide ES-C18 column, 2.1 × 150 mm, 2.7 µm. MS/MS was scanned in m/z ranges depending on the m/z values of the precursor ions. MS/MS fragmentation was performed in either CID or ETD mode. CID experiments were done with trap fragmentation. Normalized fragmentation energy was set at 35% for CID fragmentation and 35% for ETD fragmentation. MS2 data was manually interpreted.
Alkaline phosphatase treatment
Ten ug of mAb protein in AEX strip fraction were diluted in 50 uL phosphatase reaction buffer (CutSmart Buffer from New England Biolabs, Cat# B7204S, 50 mM Potassium Acetate 20 mM Tris-acetate 10 mM Magnesium Acetate 100 μg/ml BSA pH 7.9). 1 uL (10 unit) alkaline phosphatase from calf intestinal (Cat# M0290S, New England Biolabs, Ipswich, MA) was added to the sample, then the solution was incubated at 37°C for 1 hour. 10 ug chicken ovalbumin (Cat# S7951, Sigma) was also diluted in phosphatase reaction buffer (CutSmart Buffer, 50 uL), treated by 1 uL alkaline phosphatase and incubated side by side as a positive control. 10 uL solution (2 ug) was injected to LC/MS for mass analysis.
Western blot
Magic Mark XP™ Western Standard (Invitrogen) and specific concentrations of both mAb and control cell extracts (HEK293 whole cell extract and EGF-stimulated A431 cell lysate (Millipore)) were reduced with ß-mercaptoethanol plus heating at 95°C then resolved by Tris-glycine-based SDS-PAGE using a 4–20% gradient gel (Novex). Resolved proteins were subsequently electro-transferred onto nitrocellulose membrane and washed overnight in Tris-buffered saline plus 0.05% Tween20 (TBST) (Sigma) with rocking at 4°C. Membranes were then blocked for 1 h in Tris-buffered saline plus 1% BSA (TBS-BSA) (Sigma) at room temperature with continuous rocking. Primary antibodies (anti-sulfotyrosine/anti-tyrosine sulfation (Cat# 05–1100, lot# 2558454, Millipore) or anti-human IgG (H+L) (Cat# 309–005–082, lot# 07673 Jackson ImmunoResearch Laboratories Inc.)) were diluted into TBS-BSA and incubated with the membrane for 2 h at room temperature. After washing with TBST, HRP-conjugated secondary antibodies (goat-anti-mouse (Cat# 31437, lot# QG2061723, Thermo Scientific) or goat-anti-rabbit (Cat# 31463, lot# LK1368368, Thermo Scientific)) were diluted into 5% Non-fat milk protein plus 0.05% Tween20-phosphobuffered saline (Invitrogen) and incubated at room temperature for 1 h. After a final washing with TBST, chemiluminescence substrates (Thermo Scientific) were used for development; signals were recovered by exposure to photographic film (GE Healthcare Life Sciences) and subsequent processing. Nitrocellulose membrane stripping in between primary antibodies was done as described previously.Citation25
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Baeuerle PA, Huttner WB. J Cell Biol 1987; 105:2655-64; PMID:3121635; https://doi.org/10.1083/jcb.105.6.2655
- Moore KL. Proc Natl Acad Sci 2009; 106(35):14741-2; PMID:19717448; https://doi.org/10.1073/pnas.0908376106
- Monigatti F, Hekking B, Steen H. Biochim Biophys Acta 2006; 1764:1904-13; PMID:16952486; https://doi.org/10.1016/j.bbapap.2006.07.002
- Kehoe JW, Bertozzi CR. Chem Biol 2000; 7:R57-61; PMID:10712936; https://doi.org/10.1016/S1074-5521(00)00093-4
- Moore KL. J Biol Chem 2003; 278:24243-46; PMID:12730193; https://doi.org/10.1074/jbc.R300008200
- Farzan M, Mirzabekov T, Kolchinsky P, Wyatt R, Cayabyab M, Gerard NP, Gerard C, Sodroski J, Choe H. Cell 1999; 96:667-76; PMID:10089882; https://doi.org/10.1016/S0092-8674(00)80577-2
- Farzan M, Babcock GJ, Vasilieva N, Wright PL, Kiprilov E, Mirzabekov T, Choe H. J Biol Chem 2002; 277:29484-89; PMID:12034737; https://doi.org/10.1074/jbc.M203361200
- Hemmerich S, Paavola C, Bloom A, Bhakta S, Freedman R, Grunberger D, Krstenansky J, Lee S, McCarley D, Mulkins M, Wong B, Pease J, Mizoue L, Mirzadegan T, Polsky I, Thompson K, Handel TM, Jarnagin K. Biochemistry 1999; 38:13013-25; PMID:10529171; https://doi.org/10.1021/bi991029m
- Pouyani T, Seed B. Cell 1995; 83:333-43; PMID:7585950; https://doi.org/10.1016/0092-8674(95)90174-4
- Leppänen A, Mehta P, Ouyang YB, Ju T, Helin J, Moore KL, van Die I, Canfield WM, McEver RP, Cummings RD. J Biol Chem 1999; 274:24838-48; PMID:10455156; https://doi.org/10.1074/jbc.274.35.24838
- Somers WS, Tang J, Shaw GD, Camphausen RT. Cell 2000; 103:467-79; PMID:11081633; https://doi.org/10.1016/S0092-8674(00)00138-0
- Choe H, Li W, Wright PL, Vasilieva N, Venturi M, Huang CC, Grundner C, Dorfman T, Zwick MB, Wang L, Rosenberg ES, Kwong PD, Burton DR, Robinson JE, Sodroski JG, Farzan M. Cell 2003; 114:161-70; PMID:12887918; https://doi.org/10.1016/S0092-8674(03)00508-7
- Walsh G, Jefferis R. Nat Biotechnol 2006; 24:1241-52; PMID:17033665; https://doi.org/10.1038/nbt1252
- Stone SR, Hofsteenge J. Biochemistry 1986; 25:4622-8; PMID:3768302; https://doi.org/10.1021/bi00364a025
- Jefferis G, W. R. Nature Biotechnology 2006; 24:1241-52; PMID:17033665; https://doi.org/10.1038/nbt1006-1230
- Nemeth-Cawley JF, Karnik S, Rouse JC. J Mass Spectrom 2001; 36:1301-11; PMID:11754122; https://doi.org/10.1002/jms.235
- Mikesh LM, Ueberheide B, Chi A, Coon JJ, Syka JE, Shabanowitz J, Hunt DF. Biochim Biophys Acta 2006; 1764:1811-22; PMID:17118725; https://doi.org/10.1016/j.bbapap.2006.10.003
- Yu Y, Hoffhines AJ, Moore KL, Leary JA. Nat Methods 2007; 4:583-8; PMID:17558413; https://doi.org/10.1038/nmeth1056
- Xu J, D. X., Tang M, Li L, Xiao L, Yang L, Zhong J, Bode AM, Dong Z, Tao Y, Cao Y. PLoS One 2013; 8:e56114; PMID:23472069; https://doi.org/10.1371/journal.pone.0056114
- Deborah L, Grainger BH, Du Chang, Ping Lan. Cleaning up Western blot signals from immunoprecipitated samples using alternative detection methods. Biotechniques 2014; 56:149-50; https://doi.org/10.2144/000114149
- Hortin G, Folz R, Gordon JI, Strauss AW. Biochem Biophys Res Commun 1986; 141:326-33; PMID:3801003; https://doi.org/10.1016/S0006-291X(86)80372-2
- Rosenquist GL, Nicholas HB Jr. Protein Sci 1993; 2:215-22; PMID:8443599; https://doi.org/10.1002/pro.5560020210
- Teramoto T1, Fujikawa Y, Kawaguchi Y, Kurogi K, Soejima M, Adachi R, Nakanishi Y, Mishiro-Sato E, Liu MC, Sakakibara Y, Suiko M, Kimura M, Kakuta Y. Nat Commun 2013; 4:1572; PMID:23481380; https://doi.org/10.1038/ncomms2593
- Monigatti F, Gasteiger E, Bairoch A, Jung E. Bioinformatics 2002; 18:769-70; PMID:12050077; https://doi.org/10.1093/bioinformatics/18.5.769
- Kaufmann SH, Ewing CM, Shaper JH. Anal Biochem 1987; 161:89-95; PMID:3578791; https://doi.org/10.1016/0003-2697(87)90656-7