
ABSTRACT
Sequence variant analysis (SVA) is critical in therapeutic protein development because it ensures the absence of genetic mutations of a production clone or high-level misincorporations during cell culture. While software for searching sequence variants from mass spectrometry data are available, effectively distinguishing true positives from a large number of false positives in the reported hits or identifications found in the error tolerant search mode is a challenge. This verification process must be done manually and can take several days or even weeks to accomplish. We report here the use of a Perl-based script to evaluate every identified hit to remove the false positives from the search results of PepFinder™ (also known as MassAnalyzer) based on orthogonal criteria. Our data show that the false positives from PepFinder™ output were reduced ∼4-fold without loss of accuracy in the detection of true identifications, representing a more than 70% reduction in time compared with the manual data verification process.
Abbreviations
| LC-MS/MS | = | Liquid chromatography coupled with tandem mass spectrometry |
| mAb | = | Monoclonal antibody |
| SVA | = | Sequence variant analysis |
Introduction
Sequence variants are erroneous amino acid substitutions in the primary structure of proteins. During therapeutic protein development, sequence variants may cause protein misfolding, aggregation, and eventually affect drug safety and efficacy; thus, early detection of these species is of great importance in product and process development. Depending on the origin, sequence variants can be classified into 2 categories, mutations and misincorporations. Mutations are DNA-level errors that are most commonly introduced by incorrect nucleotide incorporations during DNA replications. Though the mutation rate in mammalian cell culture is fairly low (10−8–10−6), these errors will be carried by all the progeny of that cell, and the transfection and gene amplification steps in therapeutic protein development can increase the chance of mutation, thereby posing a potential risk for the cell line stability.Citation1,Citation2 In contrast, misincorporations are due to incorrect mRNA or amino acid incorporations during transcription (DNA to mRNA) or translation (mRNA to protein) stages. It is estimated that the rate of misincorporation is around 10−5 to 10−3.Citation3,Citation4,Citation5 Zhang discussed the potential causes for misincorporations under balanced feed condition and concluded that misincorporations are not random but normally involve only a single-base change between the codons of corresponding amino acids.Citation6 As highlighted by Zhang, the main causes are “ G(mRNA)/U(tRNA) mismatches at any of the 3 codon positions and certain additional wobble position mismatches (C/U and/or U/U)." Most of the commonly seen misincorporations, such as serine to asparagine (S->N) and valine to isoleucine (V->I), could be explained by this model. Though misincorporations naturally occur at very low levels, the unusual increase of a certain kind of misincorporation can be an indicator of a non-optimized process, e.g., the starvation of certain amino acids during cell culture.Citation7,Citation8
While mutations can be detected at both DNA and protein levels, amino acid misincorporation (sequence variants) can only be detected at the protein level. Tandem mass spectrometry (LC-MS/MS) is the most frequently used tool to detect sequence variants at the protein level. Though the experimental setup is nearly the same as a regular peptide map, special software must be implemented to search sequence variants because these peptides do not completely match the sequence in the database. Notable examples of these software tools include Mascot Error Tolerance Search (Matrix Science Inc.),Citation9 Byonic (Protein Matrics Inc.),Citation10 and PepFinder™ (Thermo Scientific Inc., formerly known as MassAnalyzer by Amgen).Citation11 These software products are different from traditional database search engines (e.g., Sequest,Citation12 Mascot) in that they can identify unexpected mass shifts and annotate these mass shifts as modifications or sequence variants. The typical process starts with building partial peptide sequences from tandem MS spectra and then detecting mass shifts by comparing precursor masses. However, due to the numerous probabilities and combinations, the chances of random matches are extremely high compared with regular database search, making sequence variants identifications error-prone. For instance, in one study, of the 7 sequence variants reported in Mascot Error Tolerance Search, only 1 turned out to be correct after manual assessment.Citation13 Nonetheless, manual verification is an inevitable and time-consuming step to authenticate the results in nearly all published works.Citation13-15 Alternative methods such as peak alignment have been exploited to increase efficiency, but the false positive rate is still very high and requires one-by-one verifications.Citation15 For a typical sequence variant analysis experiment, sample preparation, running the instruments and software search may only take 2-3 days, but it may take days or even weeks to manually verify the identifications by checking various criteria, such as retention time, mass accuracy, and the MS/MS spectra.
In our study, we used PepFinder™, a new commercial software specifically designed for characterization of biologics, to perform sequence variant searches. Similar to other software, the identification process is realized by comparing a precursor mass with all the theoretical peptide masses from the protein, then checking MS/MS to determine whether the mass difference can be located to a certain residue and whether it corresponds to a known modification or amino acid replacement. Here, we report the use of a Perl script developed in-house to automatically evaluate each identification to remove false positives based on orthogonal criteria to minimize manual effort and facilitate the data analysis process. These previously unavailable criteria can be extracted and an empirically determined threshold can be applied to quickly filter out identifications that are out of the target range. The script is not a search engine, but instead it is intended to filter PepFinder™ results to minimize manual verification rather than discover new sequence variants. A LC-MS/MS experiment using a known protein as a spiking agent was performed to prove the effectiveness of the script.
Results
Spiking one monoclonal antibody (mAb) into a similar mAb is one of the most definitive ways to mimic sequence variants and test the accuracy of the post-processing script. This experimental approach maximizes confidence in whether the identified sequence variant is true or false. In contrast, for nearly all other data sets, true and false are defined by “manual verification” from analysts, and thus tend to be subjective and error-prone. In designing the spiking experiment, 7 peptides that differ by only a single amino acid were identified in the mAbs A and B. is an overview of the data processing workflow. These peptides can mimic potential sequence variants. The peptides and their corresponding identifications are shown in . Five of 7 were identified by PepFinder™ and all 5 identifications were retained after filtering with post-processing script. S40A and A341T are missed because their parent peptides are too short (5 and 2 residues, respectively) and elute too early to be detected. Reliable identification for such small peptides can also be challenging for any search engine. These peptides usually require a second enzyme digestion for full characterization.
Figure 1. Data processing workflow with (bottom) and without (top) data post-processing. Typically each experiment can identify more than one hundred mutations / misincorporations. The analyst need to examine those identifications one by one which will take a couple days or even weeks.
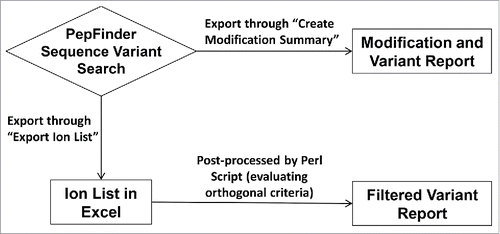
Table 1. Identification of expected variants in the spiking experiment (mAb B into mAb A, all spike levels combined). S40A and A341T are missed because their parent peptides are too short (5 and 2 residues respectively) and elute too early to be detected. Reliable identification for such small peptides can also be challenging for any search engine.
In the spiked samples, it was assumed that any matches to the expected spike peptides () and commonly known misincorporations (the misincorporations seen in literature and other molecules in Amgen) are true identifications. demonstrates the effectiveness of the filtering script. The highlighted yellow rows are the spiked peptides from mAb B and the green ones are the commonly seen misincorporations as elucidated by literature,Citation6 including S to N, S to R, N to S, V to I and A to T. Based on these criteria, the PepFinder™ output provided 57 potential sequence variants with 21 true positives, which resulted in a 63% false positive rate (FPR). A subsequent implementation of the script screened the identifications using the orthogonal criteria mentioned above to reduce the number to 30 identifications compared with 57, with 21 true positives and a FPR 29%.
Figure 2. Comparison of the identification list before (left) and after (right) the post-processing filtering. Each row is an identification from the search engine which would require ∼30 minutes to manually verify. The following criteria were used to filter the results: only retain single base substitutions, strict enzymatic site, RT score 0 to 5, % of Largest Native peak set to 50%.
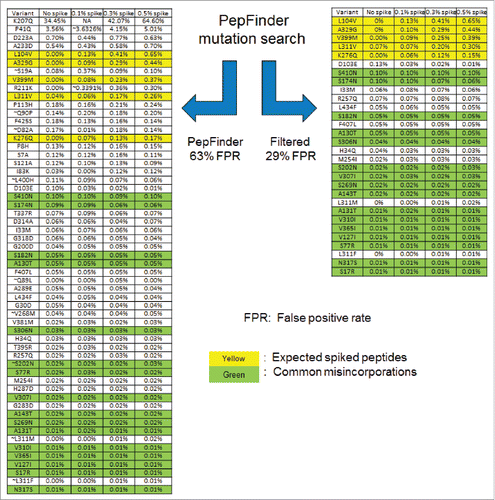
In the example above, the PepFinder™ identification list contains 21 true positives, which is the same as the filtered results. It clearly demonstrates that the filtering script could identify the false positives while retaining true positives. As mentioned, the criteria used by the script requires pre-set thresholds (e.g., RT_score, unmodified parent peak rank), and as the thresholds become tighter, the script will remove more hits, but the chance of accidentally removing true positives will also increase. To avoid this situation, the recommended threshold settings in the method section are quite relaxed and have been tested on many molecules to ensure no true positives will be accidentally removed.
shows an example of the relevant properties the script reported for S to N variant, which is one of the most common type of misincorporations. The “parent peak area” column indicates that all 6 S to N variants are detected in high abundant parent peaks (> 107), which increases the confidence of detecting them at such low levels (< 0.1%); each of their parent peptides is also the dominating form (“parent peak rank” column 100%), indicating that they are not detected in mis-cleaved or partial cleaved peptides; all the RT_scores are positive, meaning that there are retention time shifts and the directions of these shifts are in agreement with algorithm predictions. Because a larger RT_score indicates larger prediction error, most of the prediction errors are small except S269N, which has a RT_score of 4.31. The large error is due to the fact that S269N has a unique 6.5-minute retention time shift, which is significantly larger than other observed S to N variants. In fact, this S269N misincorporation has been observed in many other mAb molecules and consistently shows large retention time shift. It represents the most extreme prediction we have observed, and it is one of the reasons why a very relaxed 0 to 5 threshold was applied.
Table 2. S to N misincorporations and their retention time properties.
Another interesting phenomenon observed for these S to N misincorporations is that, although all S to N events have a good RT_score (0 < score < 5), S306N elutes later than the unmodified parent peptide while all other S to N elute earlier. This result cannot be explained by hydrophobicity alone. The good score indicates that this unusual shift is expected by the program, which takes into account not only hydrophobicity but also higher order structures. If there is no script to screen this data, other than to manually verify these sequence variants, the S306N will cause an alarm and might be labeled as a false positive. This example demonstrates the accuracy of the retention time model and its importance in data analysis.
High abundant sequence variants are cause for further investigation. To find a balance between assay sensitivity and manual effort, pre-set investigation thresholds such as > 0.3% level of variant would be manually investigated. In this case, the script is advantageous because most of the false positives tend be present in high abundance and most true misincorporations are present in low abundance. It is very clear from that, as relative abundance decreases, the number of true positive increases.
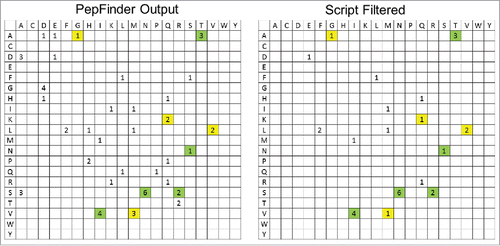
Another major application for this script is the screening of unusual misincorporations, which are usually indicative of problematic cell culture media conditions, such as starvation of certain amino acids. As shown in the results presented, misincorporations are commonly seen, but the levels are unusually or extremely low. Typically, one does not manually verify identifications at low levels due to the amount of time required. Fortunately, misincorporations are not isolated events, and the same type of misincorporations can usually be observed repeatedly on many different sites. In the spike experiment the S to N change occurred at 6 sites, and V to I at 4 sites, indicating that these are misincorporations. To some extent one can assume if a certain type of variant is identified at multiple sites, it is likely to be true. However, there is a chance that a false positive can be identified multiple times, such as misidentifying common in-source oxidation products as L to Q or carboxymethylation product as G to D. The advantage of the script is obvious for these cases: the repeated variants after filtering are more likely to be true misincorporations. displays frequency charts comparing the variants found with and without the filtering script. Each cell represents a mutation or misincorporation from the residue list in the left column to the residue list in the top row. The higher the number in each box represents a higher frequency of that event throughout the molecule. After filtering, all 5 known spikes are retained and all high numbers in the filtered chart are found to be the commonly observed misincorporations. In contrast, PepFinder™ output has several other high numbers including D to A, G to D and S to A. We manually verified and found that these 3 types corresponded to in-source C2H4O loss, non-specific carboxymethylation and in-source H2O/NH3 loss, respectively. Simply looking at the filtered frequency chart can give us a much clear and reliable view of the possible misincorporations.
Figure 3. Frequency charts showing the distribution of sequence variants by type in the PepFinder™ output (left) and script filtered results (right) of the spiking experiment (mAb B into mAb A, all spike levels combined). Each cell represents a mutation/misincorporation from the residue in the left column to the residue in the top row. Yellow colored are the expected spikes and the green ones are commonly known misincorporations.Citation6
In a real sequence variant project (), this post-processing script was applied and the threshold for manual verification was set to above 0.3%. As a result of the longer gradients and multiple injections used, the identification list for this sample was much longer compared with the spiked experiment. With PepFinder™ alone, 34 identifications needed to be manually verified when the reporting threshold is set at 0.3%, but after the filtering only 4 were left for manual verification: 2 W to R variants, one K to N variant and one H to P variant. The reduction in manual verification corresponded to ∼88% time savings. The manual identifications of the 4 variants above 0.3% determined all to be false positives. First, the 2 W to R variants are located on short 3-residue peptides and no MS/MS information was possible from the trypsin map. The identification of the W to R variants were confirmed as false positives by a second peptide map (AspN digestion). Next, the K to N variant was determined to be an un-alkylated cysteine with in-source C2H4O addition (likely from residual ethylene oxide).Citation19 Lastly, the H to P variant was determined to be unalkylated cysteine plus in-source oxidation (+16 Da), with incorrect assignment of monoisotopic peak to account for additional +2 Da shift.
Table 3. Results for a real sequence variant analysis application.
Discussion
The major advantage of using the post-processing script is time savings. For the search engines, the search for sequence variants typically results in a large number of false positives due to the numerous probabilities and combinations considered. At a very low level, MS/MS signals are usually weak, and therefore it is difficult to locate the exact position of the mass shift. The identified sequence variants must be individually and manually verified to eliminate the chance of a false positive. Typically, sample preparation by enzymatic digestion and mass spectrometry analysis can be done in 1 to 2 days, but verifying the identified variants can require at least several days, even by an experienced analyst. Therefore, the use of the post-processing script is aimed at substantially simplifying the verification process.
As demonstrated using the spiked samples, the filtering process reduced the number of identifications from 57 to 30. It should be noted that because of the intention to retain all true positives with relaxed threshold, some false positives will remain in the filtered list. The majority of the retained false positives belong to the following categories: 1) The sequence variant involves enzymatic cleavage site so the unmodified parent peptide will have significantly different length, making accurate RT_shift prediction impossible; 2) The variant is identified with a relatively large RT-score (e.g., above 3 but smaller than 5), these identifications are typically ambiguous and usually need further review; 3) Two or more modifications on the same peptide, which can confuse both PepFinder™ and the script. Nevertheless, because many criteria were automatically checked and found by the script, the analyst can bypass those criteria and focus on the verification of other possible indicators, such as supplemental proof from the digests with another enzyme. To break down the time involved, assuming it takes 30 minutes to manually verify each identification from the PepFinder™ output and 15 minutes to verify each identification from the filtered results, this lower number of identifications translated to a time-savings of 21 hours or a 76% reduction in processing time with a very low chance of missing a true positive. This time saving can significantly speed up the clone screening process as well as drug development timelines. In addition, the report will be much less dependent on an analyst's subjective judgment, thus increasing the accuracy of the assay.
The current peptide identification search engines strongly rely on MS/MS without considering any orthogonal criteria. For low abundance sequence variants, the quality of MS/MS spectra are usually not sufficient to unambiguously locate a mass shift for a certain amino acid residue, resulting in a substantial number of false positives. In this study, we demonstrated that a post-processing script can effectively distinguish between true and false positives, thus reducing the need for manual authentication in sequence variant analysis. The application of this script within Amgen has improved sequence variant identification and added speed and efficiency during processing development. Currently, the developed script reads Excel file output from PepFinder™, but, with proper formatting, it has the potential to process the Excel outputs from any algorithms, e.g., the output from Mascot error tolerance search. In addition, a web-based sequence variant database has been built within Amgen to document the mutations and misincorporations identified within a candidate therapeutic protein. In combination with the filtering script, this database allows further unequivocal confirmation of true positives.
Methods
Protein spike experiment
Two IgG2 mAb A and B, expressed using Chinese hamster ovary cells, were produced at Amgen Inc. Test samples were prepared by spiking mAb B into the solution of the analyte mAb A at 0.1%, 0.3% and 0.5% levels. Theoretically, 7 peptides in mAb A and B differ by only one amino acid, and can be seen as sequence variants. All 7 variants could be a result of a single base change. The samples were denatured and reduced in 7.2 M guanidine hydrochloride solution with dithiothreitol, carboxymethylated with iodoacetic acid and trypsin digestion was performed at 37 C in pH 7.5 Tris-HCl buffer for 30 minutes, similarly as described by Ren et al.Citation16 After trypsin digestion, samples were run on a LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA) coupled with Acquity UPLC (Waters Corporation, Milford, MA), using a 210-minute gradient (mobile phase A: Water with 0.02% v/v trifluoroacetic acid (TFA); mobile phase B: Acetonitrile with 0.02% v/v TFA. Levels of mobile phase B increase from 0 to 40% in 180 minutes). ZORBAX 300SB-C18 column (Agilent Technologies, Santa Clara, CA) at 50°C was used for the separation. For the LTQ-Orbitrap, a resolving power of 60,000 was used for MS and collision-induced dissociation with low resolution was used for MS/MS. The top 4 most abundant peaks were selected in data-dependent mode with a dynamic exclusion time of 18 seconds.
Data processing
Data analysis was performed using PepFinder™ v.1.0 (Thermo Scientific Inc.) in mutation search mode for peaks with a minimum signal-to-noise ratio of 4. Specifically designed for LC-MS/MS applications in biopharmaceutical environment, PepFinder™ searches unmodified peptides, modified peptides, as well as peptides with mutations or misincorporations. It is similar to Mascot in error tolerance search mode.
The Excel output from PepFinder™ is then post-processed by our in-house developed Perl script, which automatically checks multiple orthogonal criteria to evaluate whether an identification is likely to be false positive. Executing the script takes only a few seconds.
Post-processing script design
Peptide identification in PepFinder™ primarily relies on precursor masses as well as MS/MS spectra, but in many cases these 2 criteria are not able to provide unambiguous identifications. The post-processing script is designed to automatically extract orthogonal peptide properties from the output of PepFinder™ to further determine whether an identification is reliable. These criteria, which are discussed in detail below, include the enzymatic cleavage site, retention time shift, unmodified parent peptide area / rank, cysteine modification status, single / multiple base mutation and common variant misidentifications.
Enzymatic cleavage site
Taking a trypsin map as an example, if a certain residue is mutated to K or R, a cleavage should be expected at the C-terminus; on the other hand, if a K or R is mutated to other residues, no cleavage should take place. These rules will be cross-checked for each identification that involves a cleavable residue.
Another aspect to be checked is called transpeptidation,Citation17 which comes from the reverse activity of trypsin. Briefly, trypsin can connect 2 peptides together, e.g., adding an R to LTADK to make a new peptide LTADKR. Software that does not consider transpeptidation will typically misidentify the later peptide as a mutation from LTADKX to LTADKR.
Retention time shift
Sequence variations typically result in retention time shift due to the changes in hydrophobicity and higher order structure. Here, we utilize a peptide retention time prediction model developed by Pacific Northwest National Laboratory, called Kangas Artificial neural network model,Citation18 to calculate the expected retention time shift after the mutation happens. In the present work, the Kangas model was revised with Perl using the original parameters. The Kangas model is designed to predict the retention time of unmodified peptides. Therefore, the retention time of the native peptide, as well as the sequence variant version of the native peptide, are both predicted using Kangas model. Then, the difference in retention time is defined as the theoretical RT shift. The theoretical shift is then compared with the experimental shift to derive an empirical retention time score (RT score). In this empirical scoring system, 0 means there is no RT shift, while a negative score means the mutated peptide is shifting against the predicted retention time change. A positive RT score represents the absolute value of prediction error, so the smaller, the better. In a 3-hour gradient, a score of 1.0 corresponds roughly to the theoretical shift and experimental shifts differing by 1.0 minute (for shorter gradient, the same score will correspond to smaller RT shift). In the rare case of a perfect prediction where the error is 0, the score will be set to 1 arbitrarily to avoid confusion with no shift event. A typical score threshold is 0 to 5 (0 not included). This threshold is fairly relaxed because it essentially allows ∼5-minute prediction error in a 3-hour gradient provided there is an RT shift and the direction of the shift is as predicted.
To avoid filtering out true positives, this scoring system and threshold have been tested on several well-characterized molecules within Amgen. In doing so, we compared the automatically filtered results with the manually verified results from multiple projects. If a known true positive was accidentally removed, we investigated the root cause and modified the threshold to retain the true positive. In this way, the above mentioned relaxed threshold was obtained after careful iteration and optimization. The threshold retains all our known true positives at the cost of retaining more false positives than a stricter threshold. Because of this threshold, some manual verification of the filtered results is still expected, but with significantly fewer candidates.
Unmodified parent peptide area/rank
The levels of sequence variants are usually very low, so typically they can only be observed when the unmodified parent peaks are present in high abundance. Because of the existence of missed cleavages, partial cleavages and all other kinds of modifications, a site can usually be detected in multiple peptide forms. If a substitution site is identified in a dominating peptide form (e.g., a fully cleaved peptide), it adds confidence of the variant identification. The script will automatically report a percentage value that is calculated as the unmodified parent peak area divided by the area of the largest peak containing the substitution site. For most of the highest confidence variant identifications, this percentage approaches 100%. For practicality, the recommended threshold is 50% to avoid filtering out true positives in some extreme cases, such as the case where a modified version of the largest peak containing the sequence variant site is not selected for MS/MS fragmentation.
Cysteine modification status
This search parameter refers to the presence or absence of missed alkylation of cysteines. Alkylation of cysteines is usually very efficient. If a peptide with sequence variant has cysteines in it, these cysteines should be alkylated.
Single/multiple base mutation
PepFinder™ has the option to search only single base mutations or multiple base mutations. If the search is done in multiple-base mutation mode, the script will label the variant as from a single base change or multiple to give more information about the variant identifications. Based on our experience, nearly all the variants observed were from single base changes. For misincorporations, the most likely change occurs on the third position of the codon.Citation6 This criterion cannot unequivocally discern true positives from false positives, but it provides valuable information about the likelihood of each variant type.
Common variant misidentifications
Some of the common variant misidentification will be labeled in the output. For instance, the G to D variant is a likely misidentification from carboxymethylation of cysteine or histidine; the V to M variant is likely from double oxidation of tryptophan or tyrosine. This criterion does not have very strong discrimination power, so the purpose is to highlight probabilities. As an example, G to D is a known low level misincorporation and it is extremely difficult to distinguish from carboxymethylation (+58 Da). The script will automatically evaluate whether there is an unmodified cysteine or histidine within 3 amino acid residue distance from the G to D variant site. If so, the G to D will be marked, and it is highly likely to be a false positive due to carboxymethylation. If a G to D misincorporation is a concern for some projects, iodoacetamide should be used for alkylation, which will create a +57 Da shift without any G to D ambiguity.
The criteria mentioned above work together as a matrix to evaluate whether a variant is true or false. The first 4 criteria have strong discrimination power; if any of them failed, the identification is highly unlikely to be true. Criteria 5 and 6 are less discriminative and are mainly used as supplemental information in the subsequent manual verification. To allow some flexibility, the user can view the whole matrix to understand why some identifications were retained while others were removed. Users can also set their own acceptance criteria such as only automatically removing the identifications that failed 2 or more criteria, while manually checking the remaining identifications.
The above criteria are selected based on the manual SVA experience of the authors, and the thresholds are determined based on the applications to more than 10 Amgen molecules that had high numbers of manually verified mutations and misincorporations. The idea is to mimic manual verification, while at the same time using a relaxed threshold to avoid aggressive filtering. Sequence variants are very low probability events and our data sets can only cover a small number of sequence variant types. However, considering the algorithms described above are universal approaches with little bias toward any sequence variant type, we assume that the script works on other variant types. The script cannot discover new sequence variants, but it optimizes the reported PepFinder™ results. Readers who wish to test the program should contact the corresponding author.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Izydor Apostol, Quanzhou Luo, Sabrina Benchaar, Zhongqi Zhang, Alain Balland, Becky Scott, Lowell Brady, and Rich Rogers for helpful discussions and testing the program.
Reference
- Li I, Fu J, Hung YT, Chu, EH. Estimation of mutation rates in cultured mammalian cells. Mutat Res 1983; 111(2):253–62; PMID:6633553; https://doi.org/10.1016/0027-5107(83)90068-4
- Boesen JJ, Niericker MJ, Dieteren N, Simons JW. How variable is a spontaneous mutation rate in cultured mammalian cells? Mutat Res 1994; 307(1):121–9; PMID:7513788; https://doi.org/10.1016/0027-5107(94)90284-4
- Parker J. Errors and alternatives in reading the universal genetic code. Microbiol Mol Biol Rev 1989; 53:273–98; PMID:2677635.
- Drummond DA, Wilke CO. The evolutionary consequences of erroneous protein synthesis. Nat Rev Genet 2009; 10:715–24; PMID:19763154; https://doi.org/10.1038/nrg2662
- Ogle JM, Ramakrishnan V. Structural insights into translational fidelity. Annu Rev Biochem 2005; 74:129–77; PMID:15952884; https://doi.org/10.1146/annurev.biochem.74.061903.155440
- Zhang Z, Shah B, Bondarenko, PV. G/U and certain wobble position mismatches as possible main causes of amino acid misincorporations. Biochemistry 2013; 52(45):8165–76; PMID:24128183; https://doi.org/10.1021/bi401002c
- Wen D, Vecchi MM, Gu S, Su L, Dolnikova J, Huang YM, Foley SF, Garber E, Pederson N, Meier W. Discovery and investigation of misincorporation of serine at asparagine positions in recombinant proteins expressed in Chinese hamster ovary cells. J Biol Chem 2009; 284:32686–94; PMID:19783658; https://doi.org/10.1074/jbc.M109.059360
- Khetan A, Huang Y, Dolnikova J, Pederson NE, Wen D, Yusuf Makagiansar H, Chen P, Ryll T. Control of misincorporation of serine for asparagine during antibody production using CHO cells. Biotechnol Bioeng 2010; 107:116–23; PMID:20506364; https://doi.org/10.1002/bit.22771
- Fenyö D, Beavis RC. A method for assessing the statistical significance of mass spectrometry-based protein identifications using general scoring schemes. Anal Chem 2003; 75(4):768–74; PMID:12622365; https://doi.org/10.1021/ac0258709
- Bern M, Kil YJ, Becker C. Byonic: Advanced peptide and protein identification software. Curr Protoc Bioinformatics 2012:13–20; PMID:22948725; https://doi.org/10.1002/0471250953.bi1320s40
- Zhang Z. Large-scale identification and quantification of covalent modifications in therapeutic proteins. Anal Chem 2009; 81(20):8354–64; PMID:19764700; https://doi.org/10.1021/ac901193n
- Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 1994; 5(11):976–89; PMID:24226387; https://doi.org/10.1016/1044-0305(94)80016-2
- Yang Y, Strahan A, Li C, Shen A, Liu H, Ouyang J, Katta V, Francissen K, Zhang B. Detecting low level sequence variants in recombinant monoclonal antibodies. MAbs 2010; 2:285–98; PMID:20400866; https://doi.org/10.4161/mabs.2.3.11718
- Que AH, Zhang B, Yang Y, Zhang J, Derfus G, Amanullah A. Sequence variant analysis using peptide mapping by LC-MS/MS. BioProcess Int 2010; 8:52–60.
- Zeck A, Regula JT, Larraillet V, Mautz B, Popp O, Göpfert U, Vollertsen UEE, Gorr IH, Koll H, Papadimitriou A. Low level sequence variant analysis of recombinant proteins: An optimized approach. PloS One 2012; 7(7):e40328; PMID:22792284; https://doi.org/10.1371/journal.pone.0040328
- Ren D, Pipes, GD, Liu D, Shih LY, Nichols AC, Treuheit MJ, Bondarenko, PV. An improved trypsin digestion method minimizes digestion-induced modifications on proteins. Anal Biochem 2009; 392(1):12–21; PMID:19457431; https://doi.org/10.1016/j.ab.2009.05.018
- Fodor, S, Zhang, Z. Rearrangement of terminal amino acid residues in peptides by protease-catalyzed intramolecular transpeptidation. Anal Biochem 2006; 356(2):282–90; PMID:16859627; https://doi.org/10.1016/j.ab.2006.06.023
- Petritis K, Kangas LJ, Yan B, Monroe ME, Strittmatter EF, Qian WJ, Adkins JN, Moore RJ, Xu Y, Lipton MS, Camp DG, Smith RD. Improved peptide elution time prediction for reversed-phase liquid chromatography-MS by incorporating peptide sequence information. Anal Chem 2006; 78(14):5026–39; PMID:16841926; https://doi.org/10.1021/ac060143p
- Chen L, Sloey C, Zhang Z, Bondarenko PV, Kim H, Ren D, Kanapuram S. Chemical modifications of therapeutic proteins induced by residual ethylene oxide. J Pharm Sci 2015; 104(2):731–9; PMID:25407640; https://doi.org/10.1002/jps.24257